Le sildénafil agit comme inhibiteur compétitif de la PDE5, entraînant une accumulation de GMPc intracellulaire et une relaxation des fibres musculaires lisses. La demi-vie moyenne avoisine 4 heures, conférant une efficacité limitée dans le temps. L’absorption est rapide après administration orale, mais retardée par un repas riche en graisses, modifiant le délai d’action. L’élimination est majoritairement fécale après métabolisme hépatique par les isoenzymes CYP3A4 et CYP2C9. Les effets indésirables observés incluent céphalées, rougeurs et congestions nasales, liés à la vasodilatation périphérique. Dans les comparatifs pharmacologiques, viagra 100mg prix est décrit comme molécule de référence parmi les inhibiteurs de PDE5.
Microsoft word - tecfidera dimethyl fumarate label_2013_0326_final_font fixed.doc
HIGHLIGHTS OF PRESCRIBING INFORMATION -------CONTRAINDICATIONS------- These highlights do not include all the information needed to use TECFIDERA safely and effectively. See full prescribing information for TECFIDERA. -------WARNINGS AND PRECAUTIONS-------
TECFIDERA may cause lymphopenia. A recent CBC should be available
TECFIDERA™ (dimethyl fumarate) delayed-release capsules, for oral
before initiating treatment with TECFIDERA. A CBC is recommended
annually, and as clinically indicated. Consider withholding treatment in
Initial U.S. Approval: 2013
patients with serious infections. (5.1, 6.1)
-------INDICATIONS AND USAGE------- -------ADVERSE REACTIONS-------
TECFIDERA is indicated for the treatment of patients with relapsing forms of
Most common adverse reactions (incidence ≥10% and ≥2% placebo) were
flushing, abdominal pain, diarrhea, and nausea (6.1)
-------DOSAGE AND ADMINISTRATION------- To report SUSPECTED ADVERSE REACTIONS, contact Biogen Idec at
Starting dose: 120 mg twice a day, orally, for 7 days (2)
1-800-456-2255 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Maintenance dose after 7 days: 240 mg twice a day, orally (2)
Swallow TECFIDERA capsules whole and intact. Do not crush, chew, or
-------USE IN SPECIFIC POPULATIONS-------
Pregnancy: based on animal data, may cause fetal harm (8.1)
Take TECFIDERA with or without food (2)
-------DOSAGE FORMS AND STRENGTHS------- See 17 for PATIENT COUNSELING INFORMATION and FDA- Delayed-release capsules: 120 mg and 240 mg (3)
approved patient labeling Revised: 03/2013 FULL PRESCRIBING INFORMATION: CONTENTS* 12 CLINICAL PHARMACOLOGY 1 INDICATIONS AND USAGE 2 DOSAGE AND ADMINISTRATION
2.2 Blood Test Prior to Initiation of Therapy
13 NONCLINICAL TOXICOLOGY 3 DOSAGE FORMS AND STRENGTHS
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
4 CONTRAINDICATIONS
13.2 Animal Toxicology and/or Pharmacology
5 WARNINGS AND PRECAUTIONS 14 CLINICAL STUDIES 16 HOW SUPPLIED/STORAGE AND HANDLING 17 PATIENT COUNSELING INFORMATION 6 ADVERSE REACTIONS
17.2 Flushing and Gastrointestinal (GI) Reactions
8 USE IN SPECIFIC POPULATIONS
* Sections or subsections omitted from the full prescribing information are not
11 DESCRIPTION FULL PRESCRIBING INFORMATION 1 INDICATIONS AND USAGE
TECFIDERA is indicated for the treatment of patients with relapsing forms of multiple sclerosis.
2 DOSAGE AND ADMINISTRATION 2.1 Dosing Information
The starting dose for TECFIDERA is 120 mg twice a day orally. After 7 days, the dose should be increased to the maintenance dose of 240 mg twice a day orally. TECFIDERA should be swallowed whole and intact. TECFIDERA should not be crushed or chewed and the capsule contents should not be sprinkled on food. TECFIDERA can be taken with or without food. Administration with food may reduce the incidence of flushing [see Clinical Pharmacology (12.3)].
2.2 Blood Test Prior to Initiation of Therapy
A recent complete blood cell count (CBC) (i.e., within 6 months) is recommended before initiation of therapy to identify patients with pre-existing low lymphocyte counts.
3 DOSAGE FORMS AND STRENGTHS
TECFIDERA is available as hard gelatin delayed-release capsules containing 120 mg or 240 mg of dimethyl fumarate. The 120 mg capsules have a green cap and white body, printed with “BG-12 120 mg” in black ink on the body. The 240 mg capsules have a green cap and a green body, printed with “BG-12 240 mg” in black ink on the body.
4 CONTRAINDICATIONS
None. 5 WARNINGS AND PRECAUTIONS
5.1 Lymphopenia TECFIDERA may decrease lymphocyte counts [see Adverse Reactions (6.1)]. In the MS placebo controlled trials, mean lymphocyte counts decreased by approximately 30% during the first year of treatment with TECFIDERA and then remained stable. Four weeks after stopping TECFIDERA, mean lymphocyte counts increased but did not return to baseline. Six percent (6%) of TECFIDERA patients and <1% of placebo patients experienced lymphocyte counts <0.5x109/L (lower limit of normal 0.91x109/L). The incidence of infections (60% vs 58%) and serious infections (2% vs 2%) was similar in patients treated with TECFIDERA or placebo, respectively. There was no increased incidence of serious infections observed in patients with lymphocyte counts <0.8x109/L or 0.5x109/L. Before initiating treatment with TECFIDERA, a recent CBC (i.e., within 6 months) should be available. A CBC is recommended annually, and as clinically indicated. Withholding treatment should be considered in patients with serious infections until the infection(s) is resolved. TECFIDERA has not been studied in patients with pre-existing low lymphocyte counts. 5.2 Flushing
TECFIDERA may cause flushing (e.g., warmth, redness, itching, and/or burning sensation). In clinical trials, 40% of TECFIDERA treated patients experienced flushing. Flushing symptoms generally began soon after initiating TECFIDERA and usually improved or resolved over time. In the majority of patients who experienced flushing, it was mild or moderate in severity. Three percent (3%) of
patients discontinued TECFIDERA for flushing and <1% had serious flushing symptoms that were not life-threatening but led to hospitalization. Administration of TECFIDERA with food may reduce the incidence of flushing.
6 ADVERSE REACTIONS
The following important adverse reactions are described elsewhere in labeling: Lymphopenia, Flushing [see Warnings and Precautions (5.1, 5.2)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The most common adverse reactions (incidence ≥10% and ≥2% more than placebo) for TECFIDERA were flushing, abdominal pain, diarrhea, and nausea.
Adverse Reactions in Placebo-Controlled Trials
In the two well-controlled studies demonstrating effectiveness, 1529 patients received TECFIDERA with an overall exposure of 2244 person-years [see Clinical Studies (14)].
The adverse reactions presented in the table below are based on safety information from 769 patients treated with TECFIDERA 240 mg twice a day and 771 placebo-treated patients.
Table 1: Adverse Reactions in Study 1 and 2 reported for TECFIDERA 240 mg BID at ≥ 2% higher incidence than placebo
TECFIDERA Blood and Lymphatic System Disorders Gastrointestinal Disorders Vascular Disorders Skin and Subcutaneous Tissue Disorders Investigations
TECFIDERA caused GI events (e.g., nausea, vomiting, diarrhea, abdominal pain, and dyspepsia). The incidence of GI events was higher early in the course of treatment (primarily in month 1) and usually decreased over time in patients treated with TECFIDERA compared with placebo. Four percent (4%) of patients treated with TECFIDERA and less than 1% of placebo patients discontinued due to gastrointestinal events. The incidence of serious GI events was 1% in patients treated with TECFIDERA.
An increased incidence of elevations of hepatic transaminases in patients treated with TECFIDERA was seen primarily during the first six months of treatment, and most patients with elevations had levels < 3 times the upper limit of normal (ULN). Elevations of alanine aminotransferase and aspartate aminotransferase to ≥ 3 times the ULN occurred in a small number of patients treated with both TECFIDERA and placebo and were balanced between groups. There were no elevations in transaminases ≥ 3 times the ULN with concomitant elevations in total bilirubin > 2 times the ULN. Discontinuations due to elevated hepatic transaminases were < 1% and were similar in patients treated with TECFIDERA or placebo.
A transient increase in mean eosinophil counts was seen during the first 2 months of therapy.
Adverse Reactions in Placebo-Controlled and Uncontrolled Studies
In placebo-controlled and uncontrolled clinical studies, a total of 2513 patients have received TECFIDERA and been followed for periods up to 4 years with an overall exposure of 4603 person-years. Approximately 1162 patients have received more than 2 years of treatment with TECFIDERA. The adverse reaction profile of TECFIDERA in the uncontrolled clinical studies was consistent with the experience in the placebo-controlled clinical trials.
8 USE IN SPECIFIC POPULATIONS 8.1 Pregnancy
There are no adequate and well-controlled studies in pregnant women. In animals, adverse effects on offspring survival, growth, sexual maturation, and neurobehavioral function were observed when dimethyl fumarate (DMF) was administered during pregnancy and lactation at clinically relevant doses. TECFIDERA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
In rats administered DMF orally (25, 100, 250 mg/kg/day) throughout organogenesis, embryofetal toxicity (reduced fetal body weight and delayed ossification) were observed at the highest dose tested. This dose also produced evidence of maternal toxicity (reduced body weight). Plasma exposure (AUC) for monomethyl fumarate (MMF), the major circulating metabolite, at the no-effect dose is approximately three times that in humans at the recommended human dose (RHD) of 480 mg/day. In rabbits administered DMF orally (25, 75, and 150 mg/kg/day) throughout organogenesis, embryolethality and decreased maternal body weight were observed at the highest dose tested. The plasma AUC for MMF at the no-effect dose is approximately 5 times that in humans at the RHD.
Oral administration of DMF (25, 100, and 250 mg/kg/day) to rats throughout organogenesis and lactation resulted in increased lethality, persistent reductions in body weight, delayed sexual maturation (male and female pups), and reduced testicular weight at the highest dose tested. Neurobehavioral impairment was observed at all doses. A no-effect dose for developmental toxicity was not identified. The lowest dose tested was associated with plasma AUC for MMF lower than that in humans at the RHD.
Pregnancy Registry There is a pregnancy registry that monitors pregnancy outcomes in women exposed to TECFIDERA during pregnancy. Encourage patients to enroll by calling 1-800-456-2255.
8.3 Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when TECFIDERA is administered to a nursing woman.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
8.5 Geriatric Use
Clinical studies of TECFIDERA did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients.
11 DESCRIPTION
TECFIDERA contains dimethyl fumarate which is also known by its chemical name, dimethyl (E) butenedioate, (C6H8O4). It has the following structure:
Dimethyl fumarate is a white to off-white powder that is highly soluble in water with a molecular mass of 144.13.
TECFIDERA is provided as hard gelatin delayed-release capsules for oral administration, containing 120 mg or 240 mg of dimethyl fumarate consisting of the following inactive ingredients: microcrystalline cellulose, silicified microcrystalline cellulose, croscarmellose sodium, talc, silica colloidal silicon dioxide, magnesium stearate, triethyl citrate, methacrylic acid copolymer - Type A, methacrylic acid copolymer dispersion, simethicone (30% emulsion), sodium lauryl sulphate, and polysorbate 80. The capsule shell, printed with black ink, contains the following inactive ingredients: gelatin, titanium dioxide, FD&C blue 1; brilliant blue FCF, yellow iron oxide and black iron oxide.
12 CLINICAL PHARMACOLOGY 12.1 Mechanism of Action
The mechanism by which dimethyl fumarate (DMF) exerts its therapeutic effect in multiple sclerosis is unknown. DMF and the metabolite, monomethyl fumarate (MMF), have been shown to activate the Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) pathway in vitro and in vivo in animals and humans. The Nrf2 pathway is involved in the cellular response to oxidative stress. MMF has been identified as a nicotinic acid receptor agonist in vitro.
12.2 Pharmacodynamics Potential to prolong the QT interval
In a placebo controlled thorough QT study performed in healthy subjects, there was no evidence that dimethyl fumarate caused QT interval prolongation of clinical significance (i.e., the upper bound of the 90% confidence interval for the largest placebo-adjusted, baseline-corrected QTc was below 10 ms). 12.3 Pharmacokinetics
After oral administration of TECFIDERA, dimethyl fumarate undergoes rapid presystemic hydrolysis by esterases and is converted to its active metabolite, monomethyl fumarate (MMF). Dimethyl fumarate is not quantifiable in plasma following oral administration of TECFIDERA. Therefore all pharmacokinetic analyses related to TECFIDERA were performed with plasma MMF concentrations. Pharmacokinetic data were obtained in subjects with multiple sclerosis and healthy volunteers.
The median Tmax of MMF is 2-2.5 hours. The peak plasma concentration (Cmax) and overall exposure (AUC) increased approximately dose proportionally in the dose range studied (120 mg to 360 mg). Following administration of TECFIDERA 240 mg twice a day with food, the mean Cmax of MMF was 1.87 mg/L and AUC was 8.21 mg.hr/L in MS patients.
A high-fat, high-calorie meal did not affect the AUC of MMF but decreased its Cmax by 40%. The Tmax was delayed from 2.0 hours to 5.5 hours. In this study, the incidence of flushing was reduced by approximately 25% in the fed state.
The apparent volume of distribution of MMF varies between 53 and 73 L in healthy subjects. Human plasma protein binding of MMF is 27-45% and independent of concentration.
In humans, dimethyl fumarate is extensively metabolized by esterases, which are ubiquitous in the gastrointestinal tract, blood, and tissues, before it reaches the systemic circulation. Further metabolism of MMF occurs through the tricarboxylic acid (TCA) cycle, with no involvement of the cytochrome P450 (CYP) system. MMF, fumaric and citric acid, and glucose are the major metabolites in plasma.
Exhalation of CO2 is the primary route of elimination, accounting for approximately 60% of the TECFIDERA dose. Renal and fecal elimination are minor routes of elimination, accounting for 16% and 1% of the dose respectively. Trace amounts of unchanged MMF were present in urine.
The terminal half-life of MMF is approximately 1 hour and no circulating MMF is present at 24 hours in the majority of individuals. Accumulation of MMF does not occur with multiple doses of TECFIDERA.
Body weight, gender, and age do not require dosage adjustment.
No studies have been conducted in subjects with hepatic or renal impairment. However, neither condition would be expected to affect exposure to MMF and therefore no dosage adjustment is necessary.
No potential drug interactions with dimethyl fumarate or MMF were identified in in vitro CYP inhibition and induction studies, or in P-glycoprotein studies. Single doses of interferon beta-1a or glatiramer acetate did not alter the pharmacokinetics of MMF. Aspirin, when administered approximately 30 minutes before TECFIDERA, did not alter the pharmacokinetics of MMF.
13 NONCLINICAL TOXICOLOGY 13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies of dimethyl fumarate (DMF) were conducted in mice and rats. In mice, oral administration of DMF (25, 75, 200, and 400 mg/kg/day) for up to two years resulted in an increase in nonglandular stomach (forestomach) and kidney tumors: squamous cell carcinomas and papillomas of the forestomach in males and females at 200 and 400 mg/kg/day; leiomyosarcomas of the forestomach at 400 mg/kg/day in males and females; renal tubular adenomas and carcinomas at 200 and 400 mg/kg/day in males; and renal tubule adenomas at 400 mg/kg/day in females. Plasma MMF exposure (AUC) at the highest dose not associated with tumors in mice (75 mg/kg/day) was similar to that in humans at the recommended human dose (RHD) of 480 mg/day.
In rats, oral administration of DMF (25, 50, 100, and 150 mg/kg/day) for up to two years resulted in increases in squamous cell carcinomas and papillomas of the forestomach at all doses tested in males and females, and in testicular interstitial (Leydig) cell adenomas at 100 and 150 mg/kg/day. Plasma MMF AUC at the lowest dose tested was lower than that in humans at the RHD.
Dimethyl fumarate (DMF) and monomethyl fumarate (MMF) were not mutagenic in the in vitro bacterial reverse mutation (Ames) assay. DMF and MMF were clastogenic in the in vitro chromosomal aberration assay in human peripheral blood lymphocytes in the absence of metabolic activation. DMF was not clastogenic in the in vivo micronucleus assay in the rat.
In male rats, oral administration of DMF (75, 250, and 375 mg/kg/day) prior to and throughout the mating period had no effect on fertility; however, increases in non-motile sperm were observed at the mid and high doses. The no-effect dose for adverse effects on sperm is similar to the recommended human dose (RHD) of 480 mg/day on a body surface area (mg/m2) basis.
In female rats, oral administration of DMF (20, 100, and 250 mg/kg/day) prior to and during mating and continuing to gestation day 7 caused disruption of the estrous cycle and increases in embryolethality at the highest dose tested. The highest dose not associated with adverse effects (100 mg/kg/day) is twice the RHD on a mg/m2 basis.
Testicular toxicity (germinal epithelial degeneration, atrophy, hypospermia, and/or hyperplasia) was observed at clinically relevant doses in mice, rats, and dogs in subchronic and chronic oral toxicity studies of DMF, and in a chronic oral toxicity study evaluating a combination of four fumaric acid esters (including DMF) in rats.
13.2 Animal Toxicology and/or Pharmacology
Kidney toxicity was observed after repeated oral administration of dimethyl fumarate (DMF) in mice, rats, dogs, and monkeys. Renal tubule epithelia regeneration, suggestive of tubule epithelial injury, was observed in all species. Renal tubular hyperplasia was observed in rats with dosing for up to two years. Cortical atrophy and interstitial fibrosis were observed in dogs and monkeys at doses above 5 mg/kg/day. In monkeys, the highest dose tested (75 mg/kg/day) was associated with single cell necrosis and multifocal and diffuse interstitial fibrosis, indicating irreversible loss of renal tissue and function. In dogs and monkeys, the 5 mg/kg/day dose was associated with plasma MMF exposures less than or similar to that in humans at the recommended human dose (RHD).
A dose-related increase in incidence and severity of retinal degeneration was observed in mice following oral administration of DMF for up to two years at doses above 75 mg/kg/day, a dose associated with plasma MMF exposure (AUC) similar to that in humans at the RHD.
14 CLINICAL STUDIES
The efficacy and safety of TECFIDERA were demonstrated in two studies (Studies 1 and 2) that evaluated TECFIDERA taken either twice or three times a day in patients with relapsing-remitting multiple sclerosis (RRMS). The starting dose for TECFIDERA was 120 mg twice or three times a day for the first 7 days, followed by an increase to 240 mg twice or three times a day. Both studies included patients who had experienced at least 1 relapse over the year preceding the trial or had a brain Magnetic Resonance Imaging (MRI) scan demonstrating at least one gadolinium-enhancing (Gd+) lesion within 6 weeks of randomization. The Expanded Disability Status Scale (EDSS) was also assessed and patients could have scores ranging from 0 to 5. Neurological evaluations were performed at baseline, every 3 months, and at the time of suspected relapse. MRI evaluations were performed at baseline, month 6, and year 1 and 2 in a subset of patients (44% in Study 1 and 48% in Study 2).
Study 1: Placebo-Controlled Trial in RRMS
Study 1 was a 2-year randomized, double-blind, placebo-controlled study in 1234 patients with RRMS. The primary endpoint was the proportion of patients relapsed at 2 years. Additional endpoints at 2 years included the number of new or newly enlarging T2 hyperintense lesions, number of new T1 hypointense lesions, number of Gd+ lesions, annualized relapse rate (ARR), and time to confirmed disability progression. Confirmed disability progression was defined as at least a 1 point increase from baseline EDSS (1.5 point increase for patients with baseline EDSS of 0) sustained for 12 weeks.
Patients were randomized to receive TECFIDERA 240 mg twice a day (n=410), TECFIDERA 240 mg three times a day (n=416), or placebo (n=408) for up to 2 years. The median age was 39 years, median time since diagnosis was 4 years, and median EDSS score at baseline was 2. The median time on study drug for all treatment arms was 96 weeks. The percentages of patients who completed 96 weeks on study drug per treatment group were 69% for patients assigned to TECFIDERA 240 mg twice a day, 69% for patients assigned to TECFIDERA 240 mg three times a day and 65% for patients assigned to placebo groups.
TECFIDERA had a statistically significant effect on all of the endpoints described above and the 240 mg three times daily dose showed no additional benefit over the TECFIDERA 240 mg twice daily dose. The results for this study (240 mg twice a day vs. placebo) are shown in Table 2 and Figure 1.
Table 2: Clinical and MRI Results of Study 1 Clinical Endpoints MRI Endpoints
Percentage of subjects with no new or newly
Figure 1: Time to 12-Week Confirmed Progression of Disability (Study 1) Study 2: Placebo-Controlled Trial in RRMS
Study 2 was a 2-year multicenter, randomized, double-blind, placebo-controlled study that also included an open-label comparator arm in patients with RRMS. The primary endpoint was the annualized relapse rate at 2 years. Additional endpoints at 2 years included
the number of new or newly enlarging T2 hyperintense lesions, number of T1 hypointense lesions, number of Gd+ lesions, proportion of patients relapsed, and time to confirmed disability progression as defined in Study 1.
Patients were randomized to receive TECFIDERA 240 mg twice a day (n=359), TECFIDERA 240 mg three times a day (n=345), an open-label comparator (n=350), or placebo (n=363) for up to 2 years. The median age was 37 years, median time since diagnosis was 3 years, and median EDSS score at baseline was 2.5. The median time on study drug for all treatment arms was 96 weeks. The percentages of patients who completed 96 weeks on study drug per treatment group were 72% for patients assigned to TECFIDERA 240 mg twice a day, 70% for patients assigned to TECFIDERA 240 mg three times a day and 64% for patients assigned to placebo groups.
TECFIDERA had a statistically significant effect on the relapse and MRI endpoints described above. There was no statistically significant effect on disability progression. The TECFIDERA 240 mg three times daily dose resulted in no additional benefit over the TECFIDERA 240 mg twice daily dose. The results for this study (240 mg twice a day vs. placebo) are shown in Table 3.
Table 3: Clinical and MRI Results of Study 2 TECFIDERA Clinical Endpoints MRI Endpoints 16 HOW SUPPLIED/STORAGE AND HANDLING
TECFIDERA is available as hard gelatin delayed-release capsules in two strengths containing either 120 mg or 240 mg of dimethyl fumarate. The green and white 120 mg capsules are printed with “BG-12 120 mg” in black ink. The green 240 mg capsules are printed with “BG-12 240 mg” in black ink. TECFIDERA is available as follows:
30-day Starter Pack, (NDC 64406-007-03):
7-day bottle 120 mg capsules, quantity 14
23-day bottle 240 mg capsules, quantity 46
7-day bottle of 14 capsules (NDC 64406-005-01)
30-day bottle of 60 capsules (NDC 64406-006-02)
Store at 15°C to 30°C (59 to 86°F). Protect the capsules from light. Store in original container. Once opened, discard bottles of TECFIDERA after 90 days.
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling (Patient Information)
17.1 Dosage
Inform patients that they will be provided two strengths of TECFIDERA when starting treatment: 120 mg capsules for the 7 day starter dose and 240 mg capsules for the maintenance dose, both to be taken twice daily. Inform patients to swallow TECFIDERA capsules whole and intact. Inform patients to not crush, chew, or sprinkle capsule contents on food. Inform patients that TECFIDERA can be taken with or without food [see Dosage and Administration (2.1)].
17.2 Flushing and Gastrointestinal (GI) Reactions
Flushing and GI reactions (abdominal pain, diarrhea, and nausea) are the most common reactions, especially at the initiation of therapy, and may decrease over time. Advise patients to contact their healthcare provider if they experience persistent and/or severe flushing or GI reactions, as taking TECFIDERA with food may help [see Adverse Reactions (6.1)].
17.3 Pregnancy and Pregnancy Registry
Instruct patients that if they are pregnant or plan to become pregnant while taking TECFIDERA they should inform their physician.
Encourage patients to enroll in the TECFIDERA Pregnancy Registry if they become pregnant while taking TECFIDERA. Advise patients to call 1-800-456-2255 for more information [see Use in Specific Populations (8.1)].17.4 Lymphocyte Counts
Inform patients that TECFIDERA may decrease lymphocyte counts. A recent blood test (i.e., within 6 months) should be available before they start therapy to identify patients with pre-existing low lymphocyte counts. Blood tests are also recommended annually, and as clinically indicated [see Warnings and Precautions (5.1), Adverse Reactions (6.1)].
41347-02 Manufactured by: Biogen Idec Inc. Cambridge, MA 02142 TECFIDERA is a trademark of Biogen Idec. 2013 Biogen Idec
Patient Information TECFIDERA™ (tek" fi de' rah) (dimethyl fumarate) delayed-release capsules What is TECFIDERA? TECFIDERA is a prescription medicine used to treat people with relapsing forms of multiple sclerosis
It is not known if TECFIDERA is safe and effective in children under 18 years of age Before taking and while you take TECFIDERA, tell your doctor if you have or have had: low white blood cell counts or an infection any other medical conditions Tell your doctor if you are: pregnant or plan to become pregnant. It is not known if TECFIDERA will harm your unborn baby.
If you become pregnant while taking TECFIDERA, talk to your doctor about enrolling in the
TECFIDERA Pregnancy Registry. You can enroll in this registry by calling 1-800-456-2255. The
purpose of this registry is to monitor the health of you and your baby.
breastfeeding or plan to breastfeed. It is not known if TECFIDERA passes into your breast milk. You and
your doctor should decide if you will take TECFIDERA or breastfeed.
taking prescription or over-the-counter medicines, vitamins, or herbal supplements How should I take TECFIDERA? Take TECFIDERA exactly as your doctor tells you to take it The recommended starting dose is one 120 mg capsule taken by mouth 2 times a day for 7 days The recommended dose after 7 days is one 240 mg capsule taken by mouth 2 times a day TECFIDERA can be taken with or without food Swallow TECFIDERA whole. Do not crush, chew, or sprinkle capsule contents on food. Protect TECFIDERA from light. You can do this by storing the capsules in their original container. Throw What are the possible side effects of TECFIDERA? TECFIDERA may cause serious side effects including: decreases in your white blood cell count The most common side effects of TECFIDERA include: flushing, redness, itching, or rash
nausea, vomiting, diarrhea, stomach pain, or indigestion Flushing and stomach problems are the most common reactions, especially at the start of therapy, and may decrease over time. Call your doctor if you have any of these symptoms and they bother you or do not go away. These are not all the possible side effects of TECFIDERA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. For more information go to dailymed.nlm.nih.gov. General Information about the safe and effective use of TECFIDERA Medicines are sometimes prescribed for purposes other than those listed in this Patient Information. Do
not use TECFIDERA for a condition for which it was not prescribed. Do not give TECFIDERA to other people, even if they have the same symptoms that you have. It may harm them.
If you would like more information, talk to your doctor or pharmacist. You can ask your doctor or
pharmacist for information about TECFIDERA that is written for healthcare professionals.
What are the ingredients in TECFIDERA? Active ingredient: dimethyl fumarate Inactive ingredients: microcrystalline cellulose, silicified microcrystalline cellulose, croscarmellose sodium, talc, silica colloidal silicon dioxide, magnesium stearate, triethyl citrate, methacrylic acid
copolymer - Type A, methacrylic acid copolymer dispersion, simethicone (30% emulsion), sodium lauryl sulphate, and polysorbate 80. Capsule Shell: gelatin, titanium dioxide, FD&C blue 1; brilliant blue FCF,
yellow iron oxide and black iron oxide. Manufactured by: Biogen Idec Inc., Cambridge, MA 02142, www.TECFIDERA.com or call 1-800-456-2255
This Patient Information has been approved by the U.S. Food and Drug Administration
PROGRAMME (PRELIMINARY) Tuesday, June 25 REGISTRATION AT NTNU, GLØSHAUGEN WELCOME RECEPTION AT THE ARCHBISHOP’S PALACE. CONCERT WITH CANTUS Wednesday, June 26 REGISTRATION Invited lectures Peter, M.G. Enzymology of Chitosan Degradation and Characterization of the OligosaccharidesDomard, A. Relation between the Cationicity of Glucosamine Residues and the Interactions involvi
Tragedia in cinque atti di William Shakespeare, scritta intorno al 1603. La prima rappresentazione documentata ebbe luogo il 1° novembre 1604 al Whitehall Palace di Londra. Titolo originale: "The Tragedy of Othello, the Moor of Venice". La trama di "Otello" fu sviluppata a partire dalla collezione di novelle "Hecatommiti" di Cinzio, seguita in maniera fedele. L'unico
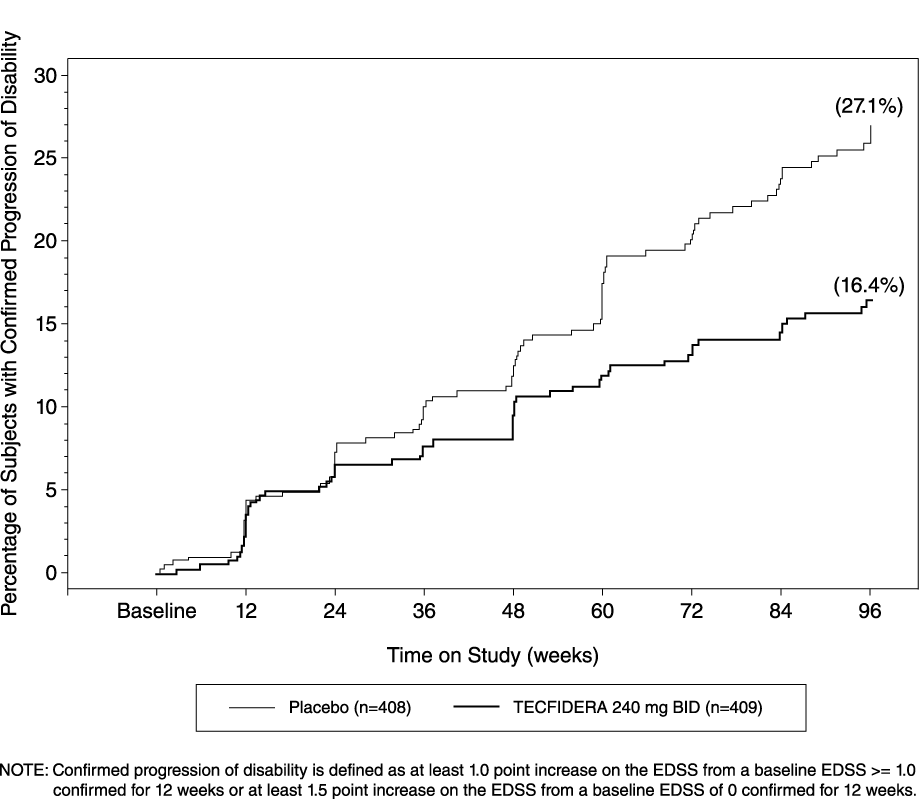 Figure 1:
Figure 1: