Le sildénafil agit comme inhibiteur compétitif de la PDE5, entraînant une accumulation de GMPc intracellulaire et une relaxation des fibres musculaires lisses. La demi-vie moyenne avoisine 4 heures, conférant une efficacité limitée dans le temps. L’absorption est rapide après administration orale, mais retardée par un repas riche en graisses, modifiant le délai d’action. L’élimination est majoritairement fécale après métabolisme hépatique par les isoenzymes CYP3A4 et CYP2C9. Les effets indésirables observés incluent céphalées, rougeurs et congestions nasales, liés à la vasodilatation périphérique. Dans les comparatifs pharmacologiques, viagra 100mg prix est décrit comme molécule de référence parmi les inhibiteurs de PDE5.
970 million druglike small molecules for virtual screening in the chemical universe database gdb-13
970 Million Druglike Small Molecules for Virtual Screening in the Chemical Universe Database GDB-13 Department of Chemistry and Biochemistry, UniVersity of Berne, Freiestrasse 3, CH-3012 Berne, Switzerland
Received March 24, 2009; E-mail: [email protected]
One of the most important chemical issues in drug discovery is
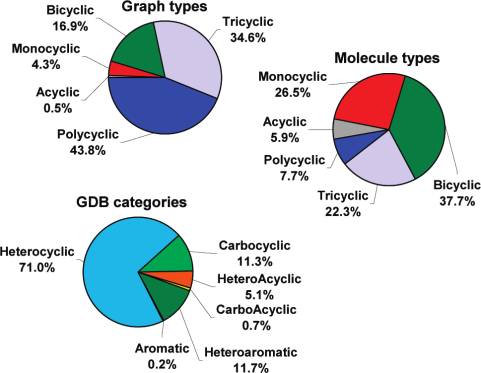
innovation, in particular at the level of small organic fragmentsthat can provide new lead structures.1 The search for novelmolecules can be assisted by in silico methods such as enumerationof chemical space,2,3 breeding of molecules by genetic algorithms,4and analysis of molecular scaffolds.5 We recently proposed anexhaustive enumeration approach for small organic molecules byassembling the chemical universe database GDB-11,6 whichdescribes the 26.4 million structures containing up to 11 atoms ofC, N, O, and F that satisfy simple chemical stability and syntheticfeasibility rules. We now report GDB-13, which enumerates in asimilar manner small organic molecules containing up to 13 atomsof C, N, O, S, and Cl. With 977 468 314 structures, GDB-13 is thelargest freely available small molecule database to date. Table 1. Structure Generation Statistics for GDB-13 Figure 1. Composition of GDB-13. Category priority: heteroaromatic >
aromatic > heterocyclic > carbocyclic > heteroacyclic (interrupted carbon
chain) > carboacyclic (continuous carbon chain).
guided drug discovery applications of GDB-11.8 Together with the
optimization of graph selection by replacing the computationally slow
MM2 minimization9 with a simple geometry-based estimation of
strained polycyclic ring systems (see the Supporting Information) and
some general code improvement, the assembly time for GDB-11 was
thus reduced 6.4-fold, from 1600 to 250 CPU h.
With these improvements, the algorithm was sufficiently fast to
910 111 673 67 356 641 39 882.08
compute the database up to 13 atoms, which produced 910 million
a Number of graph nodes considered. b Number of graphs corresponding
molecules in 40 000 CPU h (Table 1). In addition, we also produced
to saturated hydrocarbons passing topological and ring-strain criteria. c
a chlorine/sulfur set of 67.3 million compounds that enumerates
Molecules obtained from the graphs by combinatorial enumeration of
all molecules up to 13 atoms with sulfur atoms appearing in
unsaturations and heteroatoms and satisfying chemical stability andsynthetic feasibility criteria. d Molecules with a selection of Cl/S-containing
aromatic heterocycles (e.g., thiophenes), sulfones, sulfonamides,
functional groups (see the text and Supporting Information for details).
and thioureas and chlorine atoms as aromatic substituents. The Cl/S
e The database was computed in parallel on a 500-node cluster (see the
set is of interest for virtual screening because of the distinct
Supporting Information for details).
molecular shapes and functional groups that are possible with theselarger atoms.
The assembly of our previously reported GDB-11 started with a
collection of graphs7 considered as hydrocarbons, from which chemi-
The molecular diversity of GDB-13 is well-illustrated by the
cally relevant cases were selected by topological and ring-strain criteria
available molecular types (Figure 1). While polycyclic topologies
and expanded to produce more molecules by introducing unsaturations
dominate the graphs, the molecular enumeration results in a majority
and heteroatoms following valency rules.6 The limiting factor in
of monocyclic, bicyclic, and tricyclic molecules, most of which
computing GDB-11 was the elimination from this initial list of 98.4%
are heterocyclic; 54% of GDB-13 molecules have at least one three-
of unstable and/or chemically impossible molecules using functional-
or four-membered ring. The distribution of descriptor values shows
group filters. Because most of the rejected molecules contained multiple
that essentially all the molecules are druglike according to
heteroatoms, we reasoned that it might be possible to accelerate the
Lipinski10a (100%) or Vieth10b (99.5%). Many of them are also
database computation using a very fast “element-ratio” filter. Analysis
leadlike10c (98.9%) or fragmentlike10d (45.1%) (Figure 2).11
of databases of known compounds suggested cutoff values of (N +
The size of GDB-13 is a consequence of the systematic
O)/C < 1.0, N/C < 0.571, and O/C < 0.666 (see the Supporting
combinatorial enumeration. For example, between 0.2 and 18
Information). We also eliminated fluorine because it was rarely found
million compounds share the structural formula of typical marketed
and never considered in our group for synthesis in virtual-screening
drugs present in GDB-13, some of which are structurally very
8732 9 J. AM. CHEM. SOC. 2009, 131, 8732–8733 10.1021/ja902302h CCC: $40.75 2009 American Chemical Society C O M M U N I C A T I O N S
Despite these limitations, GDB-13 is to our knowledge the largest
publicly available database of virtual molecules ever reported. Itcontains a wealth of yet unknown structures to be explored andsynthesized and should provide a rich source of inspiration fordesign and synthesis in the search for new bioactive fragments notpresent in databases of already existing compounds, such as ZINC,18ACX, and PubChem. The database is available free of charge athttp://www.gdb.unibe.ch. Acknowledgment. This work was financially supported by the
University of Berne and the Swiss National Science Foundation. The authors gratefully acknowledge Chemaxon Ltd. for the donationof the academic license to the JChem package. Supporting Information Available: Details on database generation,
statistical analysis, and samples of the high-similarity structuresmentioned in Table 2. This material is available free of charge via theInternet at http://pubs.acs.org. References
(1) Bleicher, K. H.; Bo¨hm, H.-J.; Mu¨ller, K.; Alanine, A. I. Nat. ReV. DrugDiscoVery 2003, 2, 369.
(2) (a) Lederberg, W. Proc. Natl. Acad. Sci. U.S.A. 1965, 53, 134. (b) Carhart,
R. E.; Smith, D. H.; Brown, H.; Djerassi, C. J. Am. Chem. Soc. 1975, 97, 5755. (c) Benecke, C.; Grund, R.; Hohberger, R.; Kerber, A.; Laue, R.; Wieland, T. Anal. Chim. Acta 1995, 314, 141.
(3) (a) Bohacek, R. S.; McMartin, C.; Guida, W. C. Med. Res. ReV. 1996, 16,
3. (b) Ertl, P.; Jelfs, S.; Muhlbacher, J.; Schuffenhauer, A.; Selzer, P. J. Med.Figure 2. Distribution of C/N/O molecules (blue bars) and the Cl/S set Chem. 2006, 49, 4568.
(red bars) in GDB-13 according to property values. MW ) molecular weight
(4) (a) Gillet, V. J.; Myatt, G.; Zsoldos, Z.; Johnson, A. P. Perspect. Drug
in Da. TPSA ) topological polar surface area in Å2.12 clogP ) calculated
DiscoVery Des. 1995, 3, 34. (b) Globus, A.; Lawton, J.; Wipke, T.
water/octanol partition coefficient. RBC ) rotatable bond count. HBD/A
Nanotechnology 1999, 10, 290. (c) Douguet, D.; Thoreau, E.; Grassy, G. J. Comput.-Aided. Mol. Des. 2000, 14, 449. (d) Pegg, S. C.-H.; Haresco,
hydrogen-bond donor/acceptor atom count.
J. J.; Kuntz, I. D. J. Comput.-Aided. Mol. Des. 2001, 15, 911. (e) Brown, N.; McKay, B.; Gasteiger, J. J. Comput.-Aided. Mol. Des. 2004, 18, 761. Table 2. Structural Isomers of Marketed Drugs Found in GDB-13
(f) Brown, N.; McKay, B.; Gilardoni, F.; Gasteiger, J. J. Chem. Inf. Comput.Sci. 2004, 44, 1079. (g) Pierce, A. C.; Rao, G.; Bemis, G. W. J. Med. Chem. 2004, 47, 2768. (h) Lameijer, E.-W.; Kok, J. N.; Ba¨ck, T.; IJzerman, A. P. J. Chem. Inf. Model. 2006, 46, 545. (i) van Deursen, R.; Reymond,
J.-L. ChemMedChem 2007, 2, 636.
(5) (a) Koch, M. A.; Schuffenhauer, A.; Scheck, M.; Wetzel, S.; Casaulta, M.;
Odermatt, A.; Ertl, P.; Waldmann, H. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 17272. (b) Schuffenhauer, A.; Ertl, P.; Roggo, S.; Wetzel, S.; Koch,
M. A.; Waldmann, H. J. Chem. Inf. Model. 2007, 47, 47. (c) Pollock, S. N.;
Coutsias, E. A.; Wester, M. J.; Oprea, T. I. J. Chem. Inf. Model. 2008, 48,
(6) (a) Fink, T.; Bruggesser, H.; Reymond, J.-L. Angew. Chem., Int. Ed. 2005, 44, 1504. (b) Fink, T.; Reymond, J.-L. J. Chem. Inf. Model. 2007, 47, 342.
(7) McKay, B. D. Congr. Numerant. 1981, 30, 45.
(8) Nguyen, K. T.; Syed, S.; Urwyler, S.; Bertrand, S.; Bertrand, D.; Reymond,
J.-L. ChemMedChem 2008, 3, 1520.
(9) Allinger, N. L. J. Am. Chem. Soc. 1977, 99, 8127.
(10) (a) Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J. AdV. DrugDeliVery ReV. 1997, 23, 3. (b) Vieth, M.; Siegel, M. G.; Higgs, R. E.;
Watson, I. A.; Robertson, D. H.; Savin, K. A.; Durst, G. L.; Hipskind,
Common drug names as found in the DrugBank database.13
P. A. J. Med. Chem. 2004, 47, 224. (c) Teague, S. J.; Davis, A. M.; Leeson, b Number of GDB-13 molecules sharing the same structural formula.
P. D.; Oprea, T. Angew. Chem., Int. Ed. 1999, 38, 3743. (d) Congreve, c Tanimoto similarity14 compared to the parent drug; “avg” ) average
M.; Carr, R. A.; Murray, C. W.; Jhoti, H. Drug DiscoVery Today 2003, 8,
value across all compounds sharing the structural formula, “
(11) (a) Oprea, T. I. J. Comput.-Aided. Mol. Des. 2000, 14, 251. (b) Oprea,
number of these compounds with a Tanimoto value greater than 0.7.
T. I.; Allu, T. K.; Fara, D. C.; Rad, R. F.; Ostopovici, L.; Bologa, C. G. J. Comput.-Aided. Mol. Des. 2007, 21, 113.
similar to the parent compounds as estimated by their Tanimoto
(12) Ertl, P.; Rohde, B.; Selzer, P. J. Med. Chem. 2000, 43, 3714.
coefficients of structural fingerprints (Table 2).14
(13) Wishart, D. S.; Knox, C.; Guo, A. C.; Cheng, D.; Shrivastava, S.; Tzur,
D.; Gautam, B.; Hassanali, M. Nucleic Acids Res. 2008, 36, D901.
On the other hand, GDB-13 leaves out a large fraction of
(14) The standard 512-bit structural fingerprint was used: Chemical Hashed
chemical space because of the choices made to accelerate computa-
Fingerprints, www.chemaxon.com/jchem/doc/user/fingerprint.html (accessedMarch 11, 2009). For a review of chemical-similarity searching and
tion. Thus, of the 619 675 structures containing up to 13 atoms
similarity measures, see: Willett, P.; Barnard, J. M.; Downs, G. M. J. Chem.
that are found in PubChem,15 ACX,16 and the NCI Open Data-
Inf. Comput. Sci. 1998, 38, 983.
base,17 66.2% do not appear in GDB-13, either because they contain
(15) National Center for Biotechnology Information. The PubChem Project.
http://pubchem.ncbi.nlm.nih.gov (accessed Aug 4, 2008).
nonenumerated elements [e.g., F, Br, I, P, Si, metals (24.7%)] and
(16) ChemACX Ultra, version 8.0; CambridgeSoft Corporation: Cambridge, MA,
functional groups [e.g., chlorine on nonaromatic carbons, mercap-
(17) National Cancer Institute. NCI Open Database. http://cactus.nci.nih.gov
tans, sulfoxides, hemiacetals, enamines, allenes (35.9%)] or because
their heteroatom-to-carbon ratio is too high [e.g., mannitol (5.3%)]
(18) Irwin, J. J.; Shoichet, B. K. J. Chem. Inf. Model. 2005, 45, 177.
or the parent graph was not considered (0.3%). J. AM. CHEM. SOC. 9 VOL. 131, NO. 25, 2009
Understanding Indigestion and Ulcers Professor C.J. Hawkey and Dr N.J.D. WightPublished by Family Doctor Publications Limitedin association with the British Medical Association IMPORTANT This book is intended not as a substitute for personalmedical advice but as a supplement to that advice for the patient who wishes to understand more about his In particular (without limit) you should no
PROGRAMME (PRELIMINARY) Tuesday, June 25 REGISTRATION AT NTNU, GLØSHAUGEN WELCOME RECEPTION AT THE ARCHBISHOP’S PALACE. CONCERT WITH CANTUS Wednesday, June 26 REGISTRATION Invited lectures Peter, M.G. Enzymology of Chitosan Degradation and Characterization of the OligosaccharidesDomard, A. Relation between the Cationicity of Glucosamine Residues and the Interactions involvi
 970 Million Druglike Small Molecules for Virtual Screening in the Chemical
970 Million Druglike Small Molecules for Virtual Screening in the Chemical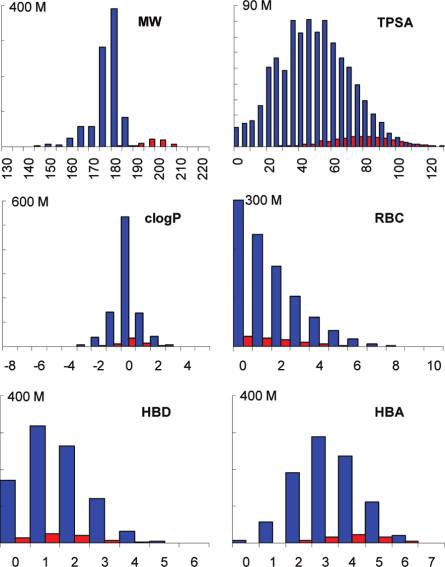 C O M M U N I C A T I O N S
C O M M U N I C A T I O N S