Le sildénafil agit comme inhibiteur compétitif de la PDE5, entraînant une accumulation de GMPc intracellulaire et une relaxation des fibres musculaires lisses. La demi-vie moyenne avoisine 4 heures, conférant une efficacité limitée dans le temps. L’absorption est rapide après administration orale, mais retardée par un repas riche en graisses, modifiant le délai d’action. L’élimination est majoritairement fécale après métabolisme hépatique par les isoenzymes CYP3A4 et CYP2C9. Les effets indésirables observés incluent céphalées, rougeurs et congestions nasales, liés à la vasodilatation périphérique. Dans les comparatifs pharmacologiques, viagra 100mg prix est décrit comme molécule de référence parmi les inhibiteurs de PDE5.
Shelton.snl.salk.edu
Annu. Rev. Physiol. 1999. 61:835–56Copyright c 1999 by Annual Reviews. All rights reservedEdwin W. McCleskeyVollum Institute, Oregon Health Sciences University, Portland, Oregon 97201-3098;e-mail: [email protected]Michael S. GoldDepartment of Oral and Craniofacial Biological Sciences, University ofMaryland–Baltimore Dental School, Baltimore, Maryland 21201;e-mail: [email protected]
pain, sensory neurons, dorsal root ganglia, Na+ channels, ligand-gated channels
Nociceptors are the first cells in the series of neurons that lead to the sensation ofpain. The essential functions of nociceptors—transducing noxious stimuli intodepolarizations that trigger action potentials, conducting the action potentialsfrom the peripheral sensory site to the synapse in the central nervous system, andconverting the action potentials into neurotransmitter release at the presynapticterminal—all depend on ion channels. This review discusses recent results inthe converging fields of nociception and ion channel biology. It focuses on (a)the capsaicin receptor and its possible role in thermosensation, (b) ATP-gatedchannels, (c) proton-gated channels, and (d ) nociceptor-specific Na+ channels.
In their landmark paper, Melzack & Wall (1) presented two opposing theorieson the nature of pain: (a) the specificity theory, which proposes that pain is asensation, like vision or hearing, conveyed via unique anatomical structures; and(b) the pattern theory, which proposes that pain results from a pattern of intenseactivity of neurons that also can encode subtle sensations such as warmth or finetouch. The specificity theory arose from the work of a number of investigators,including Charles Sherrington, who postulated the existence of cells specializedto detect noxious events and who coined the term nociceptor to describe thesecells (2). Some 50 years after Sherrington, nociceptors with sensory terminals
in skin were demonstrated unequivocally by Burgess & Perl (3) and Bessou& Perl (4). These papers reported a population of sensory neurons that weresilent until stimuli were intense enough to threaten or cause tissue damage. Incontrast, sensory neurons that detect warmth or fine touch reach maximal firingfrequency at stimulation intensities below this noxious threshold and, therefore,cannot by themselves signal the existence of tissue damage.
The modern pursuit of nociception follows several fronts. One front asks
the molecular definition of a nociceptor. Do nociceptors have unique ion chan-nels, neuropeptides, growth factor receptors, or signal transduction cascades, ascompared with other types of sensory neurons? Alternatively, nociceptors andlow-threshold sensory neurons might differ only in the level of expression ofthe same kinds of molecules. The difference is crucial to the treatment of painbecause molecules unique to nociceptors offer targets for pharmacologicallysuppressing pain without affecting other sensations. A second front asks aboutthe diversity of nociceptors. Nociceptors in skin differ from those in viscera (5),and nociceptors with unmyelinated axons differ from those with myelin (6). Athird, and very active, front is plasticity. In response to continued inflammationor nerve damage, nonnoxious stimuli can become painful (for example, the sen-sation of a warm shower on badly sunburned skin) (7). Nociceptor plasticitycontributes to this hyperalgesia, and the interaction of nerve growth factor andnociceptors is crucial (8, 9).
This review considers whether there are ion channels that are unique to no-
ciceptors. The emphasis is on a small handful of putative nociceptive channelsfor which there has been considerable recent activity: the capsaicin receptor,heat-activated channels, ATP-gated channels, proton-gated channels, and cer-tain voltage-gated Na+ channels. Other reviews consider additional channels(10, 11). The trend of this research supports the notion of nociceptor-specificion channels and, thus, encourages the pursuit of ion channels as targets foranalgesic therapy.
THE CAPSAICIN RECEPTOR AND TEMPERATURESENSATION
Capsaicin is the compound in peppers that makes them taste “hot.” Small-diameter sensory neurons selectively die after capsaicin injections in newbornrats (12), and topical application of capsaicin is analgesic because it desensitizesthe nociceptive sensory terminal (13). Because of the therapeutic potential,understanding the molecular basis of capsaicin action has been a major goal forpain research. That goal has now been reached, by Caterina et al, who success-fully cloned the receptor using a novel expression strategy (14). This landmarkpaper demonstrates that the receptor is a channel that is gated by noxious heat.
Because a vanilloid moiety is the common feature of capsaicin and resinifera-
toxin (a particularly potent ligand of the receptor), the cloned receptor is calledVR1, the first vanilloid receptor. The nucleotide sequence of VR1 predictsa protein of 838 amino acids with a molecular weight of 95,000. The pre-dicted topological organization consists of six transmembrane domains witha hydrophobic loop between the fifth and sixth domain. Noticeable sequencehomology exists between VR1 and transient receptor potential (TRP) channels,but the significance of this is unclear. Much of the homology is in ankyrin re-peats near the amino terminus, so it may only indicate a common mechanismfor cellular localization.
When expressed in either frog oocytes or a mammalian cell line, VR1 has
the appropriate pharmacological properties: activation by either capsaicin orresiniferatoxin and block by the competitive antagonist, capsazepine, and bythe noncompetitive antagonist, ruthenium red. The pore of VR1 is about tentimes more permeable to Ca2+ than to monovalent cations. Ca2+ overload likelyaccounts for the cell death seen upon chronic exposure to capsaicin.
Much of the interest in the capsaicin receptor arises from the presumption that
it has an endogenous physiological stimulus. Two results suggested that noxiousheat might be the stimulus: (a) In skin-nerve preparations, nociceptors thatrespond to heat also respond to capsaicin, whereas nociceptors that respond onlyto mechanical stimuli are insensitive to capsaicin (15); and (b) in dissociatedsensory neurons, there is an unequivocal overlap between the populations ofcells exhibiting heat-evoked current (see below) and those exhibiting capsaicin-evoked current (16). Another theory proposed that acid is the endogenousactivator of the capsaicin receptor (17). This would be relevant to pain causedby ischemia, inflammation, and infection—conditions that all drop extracellularpH in the affected tissue. Results with heterologously expressed VR1 partiallysupport both hypotheses. Caterina et al (14) show that temperatures above40◦C, which are noxious when applied to the skin, activate VR1 and that theeffect is blocked by ruthenium red. On the other hand, protons do not activateVR1, but they modulate it: The response to a submaximal dose of capsaicin canbe greatly enhanced at pH 6.3. Thus, it appears that VR1 may play a centralrole in thermosensation while also contributing to the nociceptive response totissue acidity.
A year prior to the cloning of VR1, a heat-sensitive ion channel was discov-
ered on sensory neurons (18). This channel is opened by noxious temperatures(>42◦C) and is present on about 50% of small sensory neurons but not largeones. It is absent in sympathetic neurons (19). The channel is modulated bybradykinin (18) and by prostaglandin E2 (19), two compounds that are releasedduring inflammation. Is this channel the same as VR1? In support of this no-tion, Kirschstein et al (16) show that heat-evoked currents and capsaicin-evoked
currents appear on nearly identical subsets of sensory neurons and that priorcapsaicin enhances responses to a heat stimulus. In conflict, Reichling & Levine(19) report that the heat-evoked current is not blocked by ruthenium red or cap-sazepine, as is VR1. Also, there is a quantitative discrepancy between thereported Ca2+ permeability (P) of VR1 (P
0.78) (18). Given these conflicts, it is premature to
conclude that VR1 is the heat-sensitive ion channel.
Capsaicin evokes several currents that differ in kinetics and pharmacology,
including sensitivity to capsazepine (20). Thus, VR1 will likely be one of avariety of related channels. There also appear to be differences between the heat-evoked currents reported by different laboratories. Understanding the molecularvariety and the relations between the capsaicin- and heat-evoked currents willlikely be the critical step in understanding how we sense warm temperatures. This field will progress rapidly in the next few years.
The recent molecular characterization of ATP-gated ion channels has resur-rected interest in the role of extracellular ATP in pain. The subject traces tothe mid-1970s, when Bleehen & Keele (21) proposed that ATP released fromdamaged cells contributes to pain caused by tissue damage. Two types of ex-periments suggested the hypothesis. First, ATP applied to blisters causes pain(21). Second, and more interesting, fractions of cell cytosol caused pain whenapplied to blisters (22). Most fractions had no effect, but the fractions thatcaused pain were those enriched in adenine nucleotides, the bulk of which isATP. Despite this provocative start, an unequivocal function for ATP in painremains to be described. The molecular and cellular work described belowfinds that an ATP-gated channel that is unique to nociceptors is strategicallyexpressed on sensory endings and presynaptic terminals. This raises the needfor further whole animal research to test possible roles for ATP in pain.
Two kinds of molecules must exist for ATP to function as an extracellular
mediator. The first is an ATP sensor. In the 1980s, ion channels that are openedby extracellular ATP were discovered and characterized on sensory neurons(23–25). They open in response to micromolar ATP concentrations; becausemillimolar concentrations exist within cell cytosol, substantial dilution of intra-cellular contents in extracellular space should still activate the channels. Thesecond necessary molecule must either remove or degrade ATP, thereby keep-ing extracellular concentrations low unless there is recent, nearby ATP release. This is accomplished by extracellular nucleotidases, which remove phosphategroups from ATP and are expressed in all tissues (26). Ecto-nucleotidases arefast: In brain slices, adenosine appears with a half-time of 200 ms after releaseof ATP (27). (This provides a lower estimate of the termination of ATP signals
because the adenosine arises upon removal of all three phosphates, whereas theremoval of one is sufficient to halt activation of ATP-gated channels.) Althougha bolus of extracellular ATP can, itself, cause only a transient effect, the subse-quent appearance of adenosine has been shown to inhibit synaptic transmissionin the spinal cord (28, 29).
The ion channels opened by ATP are called P2X receptors, and the G-protein
coupled receptors activated by ATP are called P2Y (30). The first P2X receptorwas cloned in 1994 (31, 32), and the P2X family rapidly expanded to sevenmembers in the next few years (33). The third member, P2X3, was cloned fromsensory neuron libraries (34, 35). Sensory neurons express mRNA for six ofthe seven P2X receptors, but in rats P2X3 mRNA is present only in sensoryneurons (34, 35). [Humans are different: P2X3 mRNA is present in humanspinal cord and heart (36).] The discovery of an ATP-gated channel uniqueto sensory neurons prompted Burnstock & Wood (37, 38) to recall and expandupon the Bleehen & Keele hypothesis (21).
Cook et al (39) pursued the suggestion that P2X3 receptors were unique
to nociceptors using several new tools: antibodies to the P2X3 protein, andpreparations of dissociated, fluorescently tagged nociceptors and muscle stretchreceptors. The nociceptors were retrogradely labeled tooth pulp afferents; thestretch receptors were muscle afferents projecting to the mesencephalic nucleusof the Vth nerve. ATP-gated currents on nociceptors are typically 10-fold largerthan on stretch receptors, and there are clear kinetic and pharmacological dif-ferences (Figure 1). Nociceptors have two distinct ATP-evoked currents. Onehas the appropriate pharmacology and transient kinetics expected of channelsmade only of P2X3 subunits (P2X3 homomers). The other has the sustainedkinetics and pharmacology of channels made of combinations of P2X3 andP2X2 subunits (P2X3/P2X2 heteromers). The single type of current in stretchreceptors behaves like P2X5 homomers. Thus, P2X3 receptors underlie bothtypes of ATP-gated current in identified nociceptors and may be unique to no-ciceptors. Use of the P2X3 antibody demonstrated that the protein is presenton nociceptive sensory endings (39). Thus, P2X3 is expressed at high density,and maybe exclusively, on nociceptors and is located at the correct subcellularsite for detecting ATP in the periphery.
Vulchanova et al (40) also showed that central terminals of sensory neurons
express P2X3 receptors. Activation of presynaptic P2X receptors enhancesglutamate release from sensory neurons in culture (41), and ATP enhancesneurotransmission at the sensory synapse in spinal cord slices (28). Thus, ATPcoreleased with glutamate at sensory presynaptic terminals first enhances trans-mission and then, upon degradation to adenosine, should suppress transmission(29).
A major difficulty in the field has been the absence of selective antagonists
for P2X receptors. Suramin and PPADS block P2X1, P2X2, P2X3, and P2X5
Nociceptors contain P2X3 receptors; stretch receptors do not. (a) Recordings from a
tooth pulp nociceptor enriched in transient type of ATP-gated current. Current is desensitized by anATP application 15 s prior to the stimulus (prepuff, left column); it is activated by α,β-methylene-ATP (middle column); it is blocked by suramin (right column). This pharmacology and the rapiddesensitization kinetics confirm the channel to be a P2X3 homomer. (b) The persistent ATP-gatedcurrent in nociceptors is activated by α,β-methylene-ATP and blocked by suramin, confirming itas a P2X2/P2X3 heteromer. (c) ATP-gated currents in stretch receptors are all persistent, but notactivated by α,β-methylene-ATP, unlike the persistent current in nociceptors. (From Reference 39.)
receptors at micromolar concentrations and the other P2X receptors at substan-tially higher concentrations (30). However, they are dirty drugs that inhibita variety of molecules. A more promising antagonist has recently appeared:Trinitrophenyl-ATP (ATP-TNP) selectively inhibits P2X1 and P2X3 recep-tors and heteromeric channels that contain one of these receptors as a subunit(42). A selective agonist, α,β-methylene-ATP, activates P2X1 and P2X3 re-ceptors (and some heteromeric channels containing these receptors) at far lower
concentrations than are needed for the other receptors (30). Distinguishing P2X1and P2X3 homomeric channels has been difficult because they both desensitize(albeit at noticeably different rates) and have similar pharmacology. Rae et al(43) argue that β,γ -methylene-L-ATP activates P2X1, but not P2X3, channels;this would indicate that the transient ATP-gated current in sensory neurons isentirely due to P2X3 receptors.
Although it is intriguing that a unique molecular machine, the P2X3 receptor,
is located at the correct subcellular site (the sensory endings of nociceptors)to allow ATP to be a mediator between tissue damage and nociception, theevidence falls short of proving such a function. The field needs further wholeanimal research. However, a number of factors make such research challengingand false negatives likely. First, ecto-nucleotidases destroy ATP in the intersti-tial space in a fraction of a second (27). Therefore, crude injection into tissueshould fail to deliver ATP to its site of action. Nucleotidase-resistant analogs ofATP must be used. Second, the homomeric P2X3 channel desensitizes quickly(<100 ms) and recovers slowly (>20 min) (44). Therefore, any procedure thatdisrupts the tissue and releases ATP should render P2X3 channels insensitive toATP for some time thereafter. Third, some P2X receptors are pH sensitive. Inparticular, the P2X2/P2X3 heteromer that makes the sustained current in noci-ceptors is greatly enhanced when pH drops below 7.0 (45). Because inflamedtissue has relatively low pH, sustained ATP responses may manifest themselvespreferentially in inflamed tissue. Consistent with these observations, perfusionof ATP into a blister causes pain (21) whereas injection of ATP into “normal”skin does not (46).
Despite the difficulties discussed above, there have been recent successes in
demonstrating ATP-evoked pain in whole animals. Bland-Ward & Humphrey(47) used a nucleotidase-resistant P2X agonist, α,β-methylene-ATP, to showthat activation of P2X receptor in the rat paw induces overt nociceptive behavior. This behavior is blocked by a local anesthetic, indicating that it is caused bynociceptor activation in the periphery. The behavior desensitizes followingpretreatment with the agonist whereas other nociceptive stimuli (formalin andbradykinin) remain effective, indicating receptor specificity in ATP-inducednociception. As expected given its sensitivity to nucleotidases, ATP itself hadjust a modest nociceptive effect and only at very high concentrations. Anotherstudy showed that P2X receptor activation enhances nociception induced byformalin and that the enhancement is blocked by suramin (48).
Muscle ischemia, inflammation, and local infection all evoke pain and each isaccompanied by local acidosis. Whether as a modulator of nociceptors or astheir primary drive, acidity plays a role in the nociception (49). In the case
of muscle ischemia, human pain increases perfectly in time with decreasingextracellular pH on the surface of the ischemic muscle (50). In skin, the me-chanical threshold of all unmyelinated nociceptors diminishes when pH drops,whereas low-threshold mechanosensors are unaffected by pH (51). A subset(38%) of mechano-heat–sensitive, unmyelinated nociceptors actively fire ac-tion potentials in response to decreasing pH, exhibiting a clear dose-responserelation between firing frequency and pH (51). The sensors proposed for thisresponse are acid-sensing ion channels that were first discovered by Krishtal &Pidoplichko (52, 53). The first acid-sensing ion channel (ASIC1) was recentlycloned (54), and as of this writing, three close relatives have been reported(55–57). This family likely will grow further.
The strategy that resulted in the discovery of ASIC1 illustrates a different
paradigm from that used to find the capsaicin and P2X receptors. As describedabove, capsaicin and P2X receptors were the targets of expression cloningstrategies designed to find them exclusively. In contrast, ASIC1 was clonedas part of an attempt to fully describe a family of ion channels that seemedunrelated to acid-sensing channels. The family includes the amiloride-sensitiveepithelial Na+ channel, which controls Na+ reabsorption by the kidney, anddegenerins, ion channels in nematodes that cause neuronal cell death when theyare constitutively active. The channels in this family all pass Na+ in preferenceto other ions, a property shared with acid-sensing channels of sensory neurons(53, 58). This analogy led the Lazdunski group to test whether any of thevarious novel clones they found were gated by protons when expressed infrog oocytes. This creative approach unmasked ASIC1 and demonstrated itsessential properties: It is closed at pH 7.4, it opens when pH drops below 7.0, itis Na+ selective, and it is expressed in sensory neurons (54). The three relatedacid-sensing channels are called ASIC2a, ASIC2b, and ASIC3 (in the originalpapers, these were called MDEG1, MDEG2, and DRASIC) (59).
The predicted membrane topology of ASICs and the related channels indi-
cates a large extracellular loop connecting two transmembrane domains withthe amino and carboxyl termini inside the cell (54). This is like the predictedtopology of P2X receptors even though there is no sequence homology (59). It is completely unlike the capsaicin receptor, thus disproving a previouslypopular theory that the capsaicin receptor was the acid-sensing channel.
Like the other members of the extended family of degenerins, each ASIC is
sensitive to amiloride and selective for Na+. They are all blocked by relativelyhigh concentrations (ca 100 µM) of amiloride that are well above the clini-cally relevant dose for diuresis. Selectivity for Na+ is an unusual property forligand-gated cation channels, which generally pass monovalent cations indis-criminately. It is unclear whether the Na+ selectivity of ASICs is significant orvestigial.
Which ASIC is responsible for the acid-evoked current in sensory neurons?
There will not be a single answer because sensory neurons express severalsuch currents that differ in their rates of activation and desensitization (53, 60). Bevan & Yeats (61) showed that a transient proton-activated current activatedin sensory neurons below pH 7 was followed by a sustained current seen onlywhen pH dropped further, to pH 6 and below. Although the transient currentis Na+ selective, the sustained current does not distinguish Na+ and K+. Thesimple explanation of this—that the transient, selective current and the sus-tained, nonselective current pass through different channels—may be wrong. Heterologous coexpression of ASIC3 and ASIC2b generates a current with atransient, selective component and a sustained, nonselective component muchlike that in native sensory neurons (57). The nonselective component provesthat ASIC2b and ASIC3 coassemble because the current is not seen when eitheris transfected alone. The heteromer generates either (a) a sustained, nonselec-tive current that is much larger than the sustained, selective current of ASIC3or (b) a channel that changes its selectivity during a prolonged stimulus.
Pain due to inflammation, infection, or ischemia is not transient, so a persist-
ing current must underlie the response (61, 62). The large, persisting currentthat is evoked when extracellular pH drops below 6 has been proposed to under-lie the pain of these conditions, but this presumes that such low pH is reached. This seems not to be true in muscle and cardiac ischemia. Muscle exercise ofan arm held in a tourniquet drops extracellular pH only to 7.0, and pain tracksperfectly with this pH change (50). The most extreme experimental cardiacischemia (complete cessation of arterial perfusion) causes extracellular pH todrop a full pH unit after about 30 min (63); such complete blockage of perfusionis quickly fatal in vivo, so such large pH changes should never occur in heart. Skin nociceptors that fire action potentials in response to pH changes haveactivation thresholds as high as pH 6.9, far too high to activate the persistentcomponent of acid-evoked current (51). It seems clear that persistent activationof a population of nociceptors occurs when extracellular pH drops from 7.4 to7.0. The molecular basis of this is not yet understood, but rapid progress in thefield may provide an answer soon.
Although the ionic currents involved in establishing the excitability of noci-ceptor terminals have yet to be identified, there is evidence that changes innociceptor excitability reflect modulation and/or changes in the expression ofseveral voltage- and Ca2+-dependent currents. These currents include voltage-dependent K+ (64, 65), Na+ (66–69), and Ca2+ (70–72) currents, an inwardlyrectifying nonselective cationic current (73), and Ca2+-dependent K+ currents
(74, 75). Rather than discuss the role each of these currents may play in themodulation of nociceptor excitability, we limit our discussion to a family ofvoltage-gated currents that has been the focus of a large body of research in thelast several years: voltage-gated Na+ currents (VGSCs). We suggest that thecellular processes involved in the modulation of VGSCs are likely to apply toother currents that may also contribute to changes in afferent excitability.
Several factors have contributed to a renaissance in research focused on eluci-
dating the contribution of VGSCs to changes in afferent excitability. First, thereis a long-standing appreciation of the critical role played by VGSCs in the gen-eration and conduction of neuronal action potentials. Second, there is a growingbody of evidence indicating that modulation of these currents is an endogenousmechanism used to control neuronal excitability (67, 76). Third, evidence frominjury in experimental animals (77–80) and humans (81, 82) suggests that ther-apeutic interventions with compounds known to block Na+ channels may beeffective for the treatment of hyperalgesia and pain. And fourth, molecularbiological tools that facilitate the study of VGSCs are now available.
At least three VGSCs have been described in mammalian sensory neurons
that are distinguished on the basis of pharmacological and biophysical prop-erties as well as on distribution among sensory neurons (83). Based on dataobtained with molecular probes specific for the various cloned VGSCs, themacroscopic Na+ current evoked from sensory neurons appears to reflect ac-tivation of a number of distinct ion channels (84). Nevertheless, these cur-rents are generally divided into two current types based on their sensitivity totetrodotoxin (TTX). One current is blocked by nanomolar concentrations ofTTX (the TTX-sensitive current) while resistant to TTX at concentrations ashigh as 10 µM (the TTX-resistant current) (85–88). Of note, evidence for asecond TTX-resistant Na+ current recently has been reported (89, 90). Unlikethe TTX-sensitive current, which is present in all sensory neurons (67, 85–88),expression of TTX-resistant current is largely restricted to a subpopulation ofneurons with nociceptor characteristics (67, 86).
The ratio of the two types of VGSC in a given neuron can have profound
effects on excitability (91). The ratio of these currents is not constant and maychange in milliseconds with changes in resting membrane potential. Rapidchange in the ratio of these two currents reflects, in part, differences in steady-state inactivation properties. TTX-sensitive Na+ currents are subject to steady-state inactivation at relatively hyperpolarized membrane potentials [V0.5 of
∼−65 mV (86)], whereas membrane depolarization is required to induce steady-state inactivation of TTX-resistant currents [V0.5 of ∼−39 mV (86)]. Thus,small changes in resting membrane potential have a large effect on the num-ber of TTX-sensitive channels available for activation and on the ratio of TTX-sensitive to insensitive Na+ current. Furthermore, because recovery from
inactivation occurs more rapidly in TTX-resistant Na+ currents than in theTTX-sensitive currents present in uninjured neurons, it has been proposed thatneurons with a large proportion of TTX-resistant Na+ currents would be able tofire long trains of action potentials (88). The relative contribution of differentVGSCs to action potential generation in afferent terminals is unknown. How-ever, there is a growing body of evidence indicating that in normal tissue, TTX-resistant Na+ current is critically involved in the process (41, 92, 93) (Figure 2). Acute ModulationChanges in nociceptor excitability are typically observed following injury. Themost rapidly developing changes in excitability (on the order of seconds tominutes) appear to reflect the action of mediators released at the site of injury. The suggestion that TTX-resistant Na+ currents contribute to injury-inducedincrease in nociceptor excitability is supported by several observations. First,
Voltage-gated Na+ currents (INa) present in dorsal root ganglion (DRG) neurons are
distinguished by sensitivity to tetrodotoxin (TTX) and gating properties. (Left column) Voltage-gated Na+ current evoked from a 25-µm DRG neuron during 20-ms command potentials rangingbetween −40 mV and +5 mV from a holding potential of −80 mV; the voltage clamp protocolused to evoke the current is shown beneath the current traces. (Center column) TTX-resistant(TTX-R) INa is isolated following bath application of 50 nM TTX; normalized conductance-voltagerelationship for activation and steady-state inactivation of TTX-R INa are plotted beneath the currenttraces (a 500-ms conditioning pulse was used to generate the steady-state inactivation data). (Rightcolumn) TTX-sensitive (TTX-S) INa is isolated as the difference between the current evoked beforeand after application of TTX (current evoked at −5, 0, and +5 mV has been omitted for clarity);normalized conductance-voltage relationships for activation and steady-state inactivation of TTX-S INa are plotted beneath the current traces (a 500-ms conditioning pulse was used to generatethe steady-state inactivation data). [From Reference 67, copyright (1996) National Academy ofSciences, USA.]
because the distribution of TTX-resistant Na+ current is restricted to a subpop-ulation of neurons with nociceptor properties (67), the current is in the rightpopulation of afferents. Second, it has been demonstrated (64, 67, 94) that TTX-resistant Na+ current is selectively modulated by hyperalgesic inflammatorymediators in a manner consistent with a role in sensitization and hyperalgesia. Hyperalgesic inflammatory mediators, including prostaglandin E2, serotonin,and adenosine, decrease the activation threshold, increase the rates of activa-tion and inactivation, and increase the magnitude of TTX-resistant Na+ current(67). These changes could contribute to both the decrease in threshold and theincrease in the number of action potentials evoked from a sensitized nocicep-tor. An inflammatory mediator chemically related to prostaglandin E2 that doesnot produce hyperalgesia, thromboxane B2, does not modulate TTX-resistantNa+ current (67). Third, the time course for the modulation of the currentin the cell body parallels that of prostaglandin E2–induced nociceptor sensi-tization (67, 95). And fourth, a µ-opioid that blocks hyperalgesic inflamma-tory mediator-induced hyperalgesia also blocks inflammatory mediator-inducedmodulation of TTX-resistant Na+ current (96).
The time course of the inflammatory mediator-induced modulation of TTX-
resistant Na+ current suggests that modulation of the current involves a phos-phorylation/dephosphorylation event(s). Consistent with this suggestion, it hasbeen demonstrated that prostaglandin E2–induced modulation of the currentinvolves activation of cAMP-dependent protein kinase (PKA) (64, but see 94). That is, prostaglandin E2–induced modulation of TTX-resistant Na+ currentwas blocked by PKA inhibitors and occluded following modulation of the cur-rent by forskolin and membrane permeable analogs of cAMP (64). Recentevidence suggests that TTX-resistant Na+ current also is modulated followingactivation of protein kinase C (MS Gold, unpublished observation).
Although it is possible that inflammatory mediator-induced modulation of
TTX-resistant Na+ current involves the phosphorylation of proteins associatedwith TTX-resistant Na+ channels, it has been demonstrated that other VGSCsare modulated subsequent to phosphorylation of the channel protein (97, 98). A channel encoding a TTX-resistant Na+ current has been cloned from sensoryneurons (99, 100). It has yet to be determined whether this channel is directlyphosphorylated following activation of PKA or protein kinase C (PKC) in situ. However, the deduced sequence of the cloned channel indicates the presence ofseveral consensus sites for PKA-induced phosphorylation, including two sitesthat appear to be unique to the Na+ channel subtype called PN3/SNS (100). Long-Term Changes in VGSCsIn addition to changes in nociceptor excitability associated with acute in-jury or inflammatory reactions, researchers have begun looking at changes in
nociceptor excitability in response to ongoing, or chronic, injury. Several ani-mal models of chronic injury have been developed to facilitate the identificationof underlying mechanisms of injury-induced changes in afferent excitability. Recent evidence suggests that different forms of injury are associated withunique changes in the expression and/or distribution of VGSCs. Nerve Injury: Complete TransectionAxotomy is one of the more widely studied models of nerve injury, in whichan entire peripheral nerve, typically the sciatic nerve, is transected (101). Ifthe nerve is prevented from reinvervating its original target, a neuroma forms. Over several weeks following axotomy, the neuroma becomes a site of hy-perexcitability, associated with a low mechanical threshold, prolonged after-discharges, and/or spontaneous activity (102).
Several lines of evidence suggest that changes in the expression and distri-
bution of VGSCs contribute to the increase in afferent excitability observedfollowing axotomy. First, spontaneous activity is exquisitely sensitive to com-pounds that block VGSCs (80). Second, VGSCs appear to accumulate at thesite of the neuroma (103). Third, changes in the expression of VGSCs areobserved. These include a decrease in two distinct species of mRNA encodingTTX-resistant Na+ channels [i.e. PN3/SNS (104) and NaN (90)] and an in-crease in the expression of mRNA encoding a TTX-sensitive Na+ current (braintype III). mRNA encoding brain type III channels is detectable in uninjured sen-sory neurons only during development (105). Consistent with these changesin mRNA levels, axotomy results in a decrease in density of TTX-resistantNa+ current present in the cell body of neurons giving rise to the injured axons(66, 106). An increase in the density of TTX-sensitive currents also is observedin situ (106). A more detailed analysis of the TTX-sensitive currents present insensory neurons following axotomy revealed the presence of a Na+ current thathas unique gating properties: The current recovers rapidly from inactivation(66). Thus, the hyperexcitability observed in the presence of axotomy mayreflect a shift in the ratio of TTX-resistant to TTX-sensitive Na+ current, witha low-threshold, rapidly “repriming” TTX-sensitive Na+ current becoming thedominant VGSC. Nerve Injury: Partial TransectionSeveral animal models have been developed to help identify changes in afferentexcitability that may be associated with nerve compression and/or partial tran-section (107–109). The most widely studied of these models was developedby Bennett & Xie (109) and involves the placement of loosely tied ligaturesaround the sciatic nerve. This injury is generally referred to as the chronicconstriction injury (CCI). Animals with CCI display signs of spontaneous pain
and hyperalgesia that begins to develop within hours of placing the ligaturesand reaches a peak 4–7 days later. These behavioral changes are associatedwith the development of spontaneous activity in the injured nerve arising, inpart, from within the sensory ganglia (110). That spontaneous activity is due, inpart, to changes intrinsic to the sensory neuron is suggested by the observationthat spontaneous activity is present in a subpopulation of sensory neuron cellbodies isolated from ganglia innervating the injured nerve (111, 112).
In contrast to axotomy, CCI does not appear to be associated with changes
in the expression of VGSC mRNA (68). However, CCI-induced changes inthe distribution of TTX-resistant Na+ channels have been reported (68). Usingan antibody against PN3, Novakovic et al (68) observed a decrease in PN3-like immunoreactivity in cell bodies of neurons associated with the injurednerve and an increase in immunoreactivity in axons near the site of injury. The decrease in immunoreactivity in the cell bodies was associated with nochange either in the density of TTX-resistant current when measured in the cellbodies of injured neurons in vitro or in mRNA levels. CCI-induced changesin the properties of TTX-resistant Na+ currents also have been reported (113). In a preliminary study, Kral et al (113) observed a hyperpolarizing shift inthe voltage dependence of TTX-resistant current measured in the cell body ofinjured neurons. That Kral et al (113) studied sensory neurons within 8 h ofdissociating whereas Novakovic et al (68) performed their experiments 18–24 hafter dissociating the neurons may explain why the former group was able todetect a change in the voltage dependence of TTX-resistant current. Chronic InflammationMost injuries to peripheral tissue are associated with inflammation. Stereo-typical inflammatory responses involve changes in vascular permeability, theinfiltration and activation of immunocompetent cells, and the synthesis and/orrelease of inflammatory mediators. Several of these processes may be involvedin an inflammation-induced increase in nociceptor excitability that developsover minutes and hours following injury (114). Although it is likely that anumber of the processes involved in the initiation of nociceptor sensitizationare responsible for the maintenance of sensitization in the presence of ongoingor chronic inflammation, a growing body of evidence indicates that chronicinflammation results in sustained changes in nociceptor physiology. For exam-ple, neuronal sprouting (115) and changes in gene expression in the presenceof chronic inflammation (115–118) are well documented.
To date, there are conflicting reports on the effects of chronic inflammation
and changes in the expression of VGSCs. Inflammation resulting from thecutaneous injection of carrageenan is associated with an increase in the ex-pression of mRNA encoding a TTX-resistant Na+ channel (SNS/PN3) (69).
An increase in the density of TTX-resistant Na+ current in the cell bodies ofsensory neurons innervating the site of inflammation is also observed (69). Incontrast, inflammation resulting from the injection of complete Freund’s adju-vant appears to have no effect on the level of SNS/PN3 mRNA (119). The basisfor the discrepancy between these two studies remains to be determined. Other Models of Peripheral Nerve InjuryChanges in the excitability of primary afferent nociceptors are observed in re-sponse to several other processes in addition to those described above. VGSCshave been implicated in two such processes, and these include diabetes andspinal cord transection. One of the many bodily organs that suffer in the pres-ence of untreated diabetes is the peripheral nervous system. In humans, diabeticneuropathy is often associated with burning pain in the extremities. In an animalmodel of diabetes, nociceptors become hyperresponsive (120). These changesin peripheral nerves are associated with a decrease in the expression of SNSmRNA (119) and with abnormalities in the properties of Na+ channels presentin axonal nodes of Ranvier (121).
Spinal transection results in bladder hyperreflexia and incontinence. These
changes occur as a result of the loss of the normal supraspinal pathway regulat-ing micturition. Work in animal models has revealed that the hyperreflexia isdue to changes in the afferents involved in the control of micturition. Bladder af-ferents become hyperexcitable, displaying a dramatic increase in mechanosen-sitivity (122). This hyperexcitability is associated with an increase in the densityof TTX-sensitive Na+ currents and a decrease in the density of TTX-resistantNa+ currents (123). A Mechanism for Changes in the Expression of VGSCsDifferences in the injury-induced changes in the expression of VGSCs appearto reflect differences in the availability of neurotrophic factors, in particularnerve growth factor (NGF). NGF is involved in regulating the expression ofboth TTX-resistant Na+ current and brain type III VGSC in somatic sensoryneurons (124–126). An increase in the concentration of NGF is associatedwith an increase in the expression of TTX-resistant Na+ current and vice versa(124–126). In contrast, an increase in the concentration of NGF decreases theexpression of brain type III currents (126). The peripheral target of innervationappears to be the source of NGF maintaining the density of TTX-resistant andbrain type III Na+ currents in sensory neurons in vivo. When access to thissource of NGF is interrupted, as in the case of axotomy, TTX-resistant Na+current density decreases whereas that of brain type III increases. Consistentwith this suggestion is the observation that providing an artificial source of NGFto axotomized neurons is capable of reversing the axotomy-induced decreases in
SNS mRNA (127) and TTX-resistant Na+ current (128). Conversely, becausethe concentration of NGF is increased in inflamed tissue (129), the expressionof TTX-resistant currents innervating this tissue should be increased. As notedabove, an increase in SNS mRNA and TTX-resistant Na+ currents is observedfollowing carrageenan-induced inflammation (69).
Although the differential regulation of VGSCs by NGF provides a useful
framework with which to understand differences in the changes observed insensory neurons in response to various injuries, several observations suggestthat this framework should be used with caution. First, there is at least onereport in which manipulating the concentration of NGF applied to sensory neu-rons had little effect on the expression of SNS/PN3 mRNA (119). Second, thechanges in the expression of SNS/PN3 mRNA and distribution of SNS/PN3channels observed in the presence of CCI (68) do not fit well within the NGFregulation framework. This is particularly true given that CCI is associated withinflammation at the site of injury (130). And third, the concentration of NGFincreases in hypertrophied bladders (131), whereas changes in the expressionof TTX-resistant and -sensitive currents in sensory neurons innervating hyper-trophied bladders are opposite to those predicted by an NGF-induced regulationof VGSCs. Ancillary SubunitsAlthough we limited our discussion of the modulation of VGSCs to mecha-nisms involving changes in the α-subunit of the channel protein, the propertiesof VGSCs, like other voltage- and Ca2+-dependent currents, are influencedby the presence of β-subunits. For example, the kinetics of inactivation ofbrain type III and skeletal muscle channels (SkM1) are increased, and the ac-tivation kinetics of brain type IIA channels are increased by the presence ofβ1-subunits (132). The presence of β1-subunit increases the expression ofcardiac Na+ channels (rH1/SkM2) (133). Although the influence of β-subunitson the VGSCs present in sensory neurons has yet to be investigated in detail,there is evidence that the presence of β-subunits may influence the expressionand/or biophysical properties of both TTX-sensitive (132) and TTX-resistantNa+ currents (100). Thus, changes in the expression of β-subunits in sen-sory neurons may influence the VGSCs present, consequently altering afferentexcitability.
Work in our laboratories is supported by grants from NIDA and NINDS. Visit the Annual Reviews home page at http://www.AnnualReviews.org
glion neurons. Neurosci. Lett. 231:33–
nisms: a new theory. Science 150:971–79
2. Sherrington CS. 1906. The Integrative Ac-
17. Bevan S, Geppetti P. 1994. Protons: small
tion of the Nervous System. New York:
stimulants of capsaicin-sensitive sensory
nerves. Trends Neurosci. 17:509–12
18. Cesare P, McNaughton P. 1996. A novel
afferent fibers responding specifically to
heat-activated current in nociceptive neu-
noxious stimulation of the skin. J. Phys-
rons and its sensitization by bradykinin. Proc. Natl. Acad. Sci. USA 93:15435–39
4. Bessou P, Perl ER. 1969. Response of cu-
fibers to noxious stimuli. J. Neurophysiol.
channel. Proc. Natl. Acad. Sci. USA 94:
5. Cervero F, J¨anig W. 1992. Visceral noci-
ceptors: a new world order? Trends Neu-
20. Liu L, Szallasi A, Simon SA. 1998.
6. Cooper BY, Vierck CJ Jr, Yeomans DC.
homovanillate, reveals vanilloid receptor
mechanisms of hyperalgesia. Pulm. Phar-
growth factor and nociception. Trends
22. Bleehen T, Hobbiger F, Keele CA. 1976.
Identification of algogenic substances in
human erythrocytes. J. Physiol. 62:131–
of inflammatory pain. Philos. Trans. R.
23. Bean BP. 1990. ATP-activated channels in
rat and bullfrog sensory neurons: concen-
activators of sensory neurons. Annu. Rev.
tration dependence and kinetics. J. Neu-
24. Jahr CE, Jessell TM. 1983. ATP excites a
channels in dorsal root ganglion neurons.
subpopulation of rat dorsal horn neurones.
In Sensory Neurons: Diversity, Develop-ment, and Plasticity, ed. A Scott, pp. 97–
12. Jancso G, Kiraly E, Jancso-Gabor A.
extracellular ATP in rat sensory neurons.
26. Zimmermann H. 1996. Biochemistry, lo-
mary sensory neurones. Nature 270:741–
nucleotidases in the nervous system.
13. Szallasi A, Blumberg PM. 1996. Vanilloid
receptors: new insights enhance potential
27. Dunwiddie TV, Diao L, Proctor WR.
as a therapeutic target. Pain 68:195–208
M, Rosen TA, Levine JD, Julius D. 1997.
ion channel in the pain pathway. Nature
28. Li J, Perl ER. 1995. ATP modulation of
15. Szolcsanyi J, Anton F, Reeh PW, Hand-
stantia gelatinosa. J. Neurosci. 15:3357–
capsaicin of mechano-heat sensitive noci-
29. Li J, Perl ER. 1994. Adenosine inhibition
ceptors in rat skin. Brain Res. 446:262–68
of synaptic transmission in the substantia
gelatinosa. J. Neurophysiol. 72:1611–21
30. North RA, Barnard EA. 1997. Nucleotide
receptors. Curr. Opin. Neurobiol. 7:346–
31. Brake AJ, Wagenbach MJ, Julius D. 1994.
dorsal root ganglia. Br. J. Pharmacol.
New structural motif for ligand-gated ion
channels defined by an ionotropic ATP re-
44. Cook SP, McCleskey EW. 1997. Desensi-
tors in nociceptors. Neuropharmacology
P2x receptor for extracellular ATP. Na-
45. Stoop R, Surprenant A, North RA. 1997.
33. Collo G, North RA, Kawashima E, Merlo-
ceptors. J. Neurophysiol. 78:1837–40
46. Reeh PW, Kress M. 1998. Boole’s algebra
family of ATP-gated ion channels. J. Neu-
47. Bland-Ward PA, Humphrey PP. 1997.
P2X receptor activation in the rat. Br. J.
by a subset of sensory neurons. Nature
35. Lewis C, Neidhart S, Holy C, North RA,
ception in the formalin test via activation
of a purinergic p2X receptor. Eur. J. Phar-
in sensory neurons. Nature 377:432–35
excitation and sensitization of nociceptors
in rat skin, in vitro. J. Neurosci. 15:3982–
P2X3 purinoceptor. Brain Res. Mol. Brain
50. Issberner U, Reeh PW, Steen KH. 1996.
37. Burnstock G, Wood JN. 1996. Purinergic
primary afferent neurotransmission. Curr.
38. Burnstock G. 1996. A unifying puriner-
duce lasting excitation and sensitization
gic hypothesis for the initiation of pain.
in rat skin, in vitro. J. Neurosci. 12:86–95
39. Cook SP, Vulchanova L, Hargreaves KM,
52. Krishtal OA, Pidoplichko VI. 1980. A re-
ceptor for protons in the nerve cell mem-
stretch-sensing neurons. Nature 387:505–
53. Krishtal OA, Pidoplichko VI. 1981. A re-
ceptor for protons in the membrane of sen-
terminals. Neuropharmacology 36:1229–
55. Waldmann R, Bassilana F, de Weille J,
nel specific for sensory neurons. J. Biol.
56. Bassilana F, Champigny G, Waldmann R,
nodose neurones. J. Physiol. 509:411–17
43. Rae MG, Rowan EG, Kennedy C. 1998.
erties. J. Biol. Chem. 272:28819–22
57. Lingueglia E, de Weille JR, Bassilana F,
channels in brain and dorsal root ganglion
cells. J. Biol. Chem. 272:29778–83
70. Bean BP. 1989. Neurotransmitter inhi-
a calcium component. Neurosci. Lett.
changes in channel voltage dependence.
71. McCleskey EW. 1994. Calcium channels:
gated cation channels: neuronal acid sen-
cellular roles and molecular mechanisms.
nels. Curr. Opin. Neurobiol. 8:418–24
72. Nicol GD, Klingberg DK, Vasko MR.
60. Pidoplichko VI. 1992. Ammonia and pro-
ton gated channel populations in trigemi-
nal ganglion neurons. Gen. Physiol. Bio-
substance P in avian sensory neurons. J.
61. Bevan S, Yeats J. 1991. Protons activate
73. Ingram SL, Williams JT. 1994. Opioid in-
hibition of Ih via adenylyl cyclase. Neuron
of rat dorsal root ganglion neurones. J.
74. Weinreich D. 1995. Cellular mechanisms
62. Steen KH, Issberner U, Reeh PW. 1995.
vagal sensory nerve excitability. Pulm.
nociceptor excitation. Neurosci. Lett. 199:
75. Gold MS, Shuster MJ, Levine JD. 1996.
terhyperpolarization in prostaglandin E2-
time of onset and severity of acidosis in
induced sensitization of cultured rat sen-
myocardial ischaemia. J. Mol. Cell. Car-
sory neurons. Neurosci. Lett. 205:161–64
64. England S, Bevan S, Docherty RJ.
ergic modulation of sodium current in hip-
resistant sodium current in neonaatal rat
dorsal root ganglion neurons via the cyclic
AMP-protein kinase A cascade. J. Phys-
sodium channel alpha subunit. J. Neu-
77. Abram SE, Yaksh TL. 1994. Systemic li-
sory neurons. J. Neurophysiol. 77:167–76
sensitization in the rat. Anesthesiology
78. Chabal C, Russell LC, Burchiel KJ. 1989.
active fibers originating in rat sciatic neu-
neurons after nerve injury. J. Neurosci.
evoked activity in identified primary affer-
ent fibers: systemic lidocaine suppresses
phase-2 activity. Pain 64:345–55
rent in nociceptors. Proc. Natl. Acad. Sci.
80. Devor M, Wall PD, Catalan N. 1992.
ing nerve conduction. Pain 48:261–68
oral mexiletine for the treatment of pain
pathic conditions. J. Neurosci. 18:2174–
after peripheral nerve injury. Anesthesiol-
82. Rizzo MA. 1997. Successful treatment of
ple positively to tetrodotoxin-insensitive
molecular pain mechanism. J. Neurol. Sci.
capsaicin-sensitive rat sensory neurons. J.
95. Gold MS, Dastmalchi S, Levine JD. 1996.
Co-expression of nociceptor properties in
ganglion neurons. Brain Res. 592:283–
adult rat in vitro. Neuroscience 71:265–
84. Black JA, Dib-Hajj S, McNabola K, Jeste
hibits prostaglandin E2-induced potentia-
tion of a TTX-resistant Na+ current in rat
alpha-subunit mRNAs. Brain Res. Mol.
sensory neurons in vitro. Neurosci. Lett.
root ganglion neurons. I. Sodium currents.
86. Ogata N, Tatebayashi H. 1993. Kinetic
rat dorsal root ganglia. J. Physiol. 466:9–
87. Roy ML, Narahashi T. 1992. Differential
tetrodotoxin-resistant sodium channels in
99. Akopian AN, Sivilotti L, Wood JN.
rat dorsal root ganglion neurons. J. Neu-
88. Elliott AA, Elliott JR. 1993. Characteriza-
sodium currents in small cells from adult
LM, Koch BD, Jakeman LB, et al. 1996.
rat dorsal root ganglia. J. Physiol. 463:39–
Structure and function of a novel voltage-
89. Rush AM, Elliott JR. 1997. Phenytoin and
channel specfic to sensory neurons. J.
carbamazepine: differential inhibition of
sodium currents in small cells from adult
101. Wall PD, Gutnick M. 1974. Ongoing ac-
rat dorsal root ganglia. Neurosci. Lett.
tivity in peripheral nerves: the physiology
90. Dib-Hajj SD, Tyrrell L, Black JA, Wax-
ing from a neuroma. Exp. Neurol. 43:580–
102. Devor M. 1994. The pathophysiology of
damaged peripheral nerves. In Textbook
and down-regulated after axotomy. Proc.of Pain, ed. PD Wall, R Melzack, pp. 79–
100. New York: Churchill Livingstone.
91. Schild JH, Kunze DL. 1997. Experimen-
tal and modeling study of Na+ current het-
103. Devor M, Govrin LR, Angelides K. 1993.
impact on neuronal discharge. J. Neuro-
104. Dib-Hajj S, Black JA, Felts P, Waxman
ity of rat intracranial meningeal afferents
by mechanical and chemical stimuli. Soc.
sory neurons following axotomy. Proc.
93. Jeftinija S. 1994. The role of tetrodotoxin-
Natl. Acad. Sci. USA 93:14950–54
105. Waxman SG, Kocsis JD, Black JA. 1994.
mary afferent fibers. Brain Res. 639:125–
pressed in embryonic but not adult spinal
following axotomy. J. Neurophysiol. 72:
118. Ji RR, Zhang Q, Law PY, Low HH, Elde
106. Zhang JM, Donnelly DF, Song XJ, Lam-
delta-, and kappa-opioid receptor-like im-
munoreactivities in rat dorsal root ganglia
excitability of dorsal root ganglion cells
after carrageenan-induced inflammation.
with unmyelinated axons. J. Neurophys-
diameter polyethylene cuffs applied to the
tion of expression of the sensory neuron-
rat sciatic nerve induce a painful neuropa-
matory and neuropathic pain. Mol. Cell.
sis of axonal alterations. Pain 64:37–57
108. Seltzer Z, Dubner R, Shir Y. 1990. A novel
120. Ahlgren SC, White DM, Levine JD.
behavioral model of neuropathic pain dis-
orders produced in rats by partial sciatic
the streptozotocin-diabetic rat. J. Neuro-
109. Bennett GJ, Xie YK. 1988. A peripheral
121. Brismar T. 1993. Abnormal Na-currents
orders of pain sensation like those seen in
in diabetic rat nerve nodal membrane. Di-
110. Kajander KC, Wakisaka S, Bennett GJ.
122. de Groat WC, Kawatani M, Hisamitsu T,
of a painful peripheral neuropathy in the
bladder function following spinal cord in-
jury. J. Auton. Nerv. Syst. 30(Suppl.):S71–
111. Study RE, Kral MG. 1996. Spontaneous
action potential activity in isolated dor-
123. Yoshimura N, de Groat WC. 1997. Plas-
a painful neuropathy. Pain 65:235–42
rones innervating rat urinary bladder fol-
112. Petersen M, Zhang J, Zhang JM, LaMotte
lowing spinal cord injury. J. Physiol.
ity and responses to norepinephrine in dis-
124. Helliwell RJA, Winter J, McIntyre P, Be-
sociated dorsal root ganglion cells after
van S. 1997. NGF regulates the expression
chronic nerve constriction. Pain 67:391–
nel in cultured sensory neurones. Soc.
113. Kral MG, Xiong Z, Study RE. 1997. Al-
neuropathy. Soc. Neurosci. Abstr. 23:1476
capsaicin-sensitivity in adult rat sensory
114. Levine JD, Basbaum AI, Fields HL. 1993.
CJ. 1995. Nerve growth factor contributes
to the up-regulation of growth-associated
sory neurons. NeuroReport 8:2331–35
following peripheral inflammation. Neu-
116. Cho HJ, Kim SY, Park MJ, Kim DS, Kim
growth factor in vivo. J. Neurophysiol.
the dorsal root ganglion following periph-
eral inflammation. Brain Res. 749:358–62
117. Donnerer J, Schuligoi R, Stein C, Amann
R. 1993. Upregulation, release and axonal
neous afferent neurons. J. Neurophysiol.
growth factor. Regul. Pept. 46:150–54
129. Constantinou J, Reynolds ML, Woolf CJ,
Safieh-Garabedian B, Fitzgerald M. 1994.
ronal form and function. J. Clin. Invest.
ing rat skin: upregulation following skin
Goldin AL. 1994. The adult rat brain beta
130. Wagner R, Janjigian M, Myers RR. 1998.
1 subunit modifies activation and inacti-
alpha subunits. J. Biol. Chem. 269:17649–
133. Qu Y, Isom LL, Westenbroek RE, Rogers
JC, Tanada TN, et al. 1995. Modulation of
131. Steers WD, Kolbeck S, Creedon D, Tut-
cardiac Na+ channel expression in Xeno-pus oocytes by beta 1 subunits. J. Biol.
urinary bladder of the adult regulates neu-
References on Delusional Parasitosis and Related Issues Aizenberg, D., B. Schwartz and Z. Zemishlany. 1991. Delusional parasitosis associated with phenelzine. British Journal of Psychiatry 159:716-717. Aleshire, I. 1954. Delusions of parasitosis: Report of successful care with antipellagrous treatment. Journal of the American Medical Association 155:15-17. Altschuler, D. Z., M. Crutche
Emergency Safety Guide TARATEK SB ENVIRONMENTALLY HAZARDOUS SUBSTANCE, LIQUID, N.O.S Propiconazole, Tebuconazole & Permethrin as a suspension concentrate Pale Cream Liquid Health and Environmental Hazards Environment Very toxic to fish and bees. Avoid discharge to waterways. Ingestion Inhalation May cause skin irritation & May cause respiratory Low
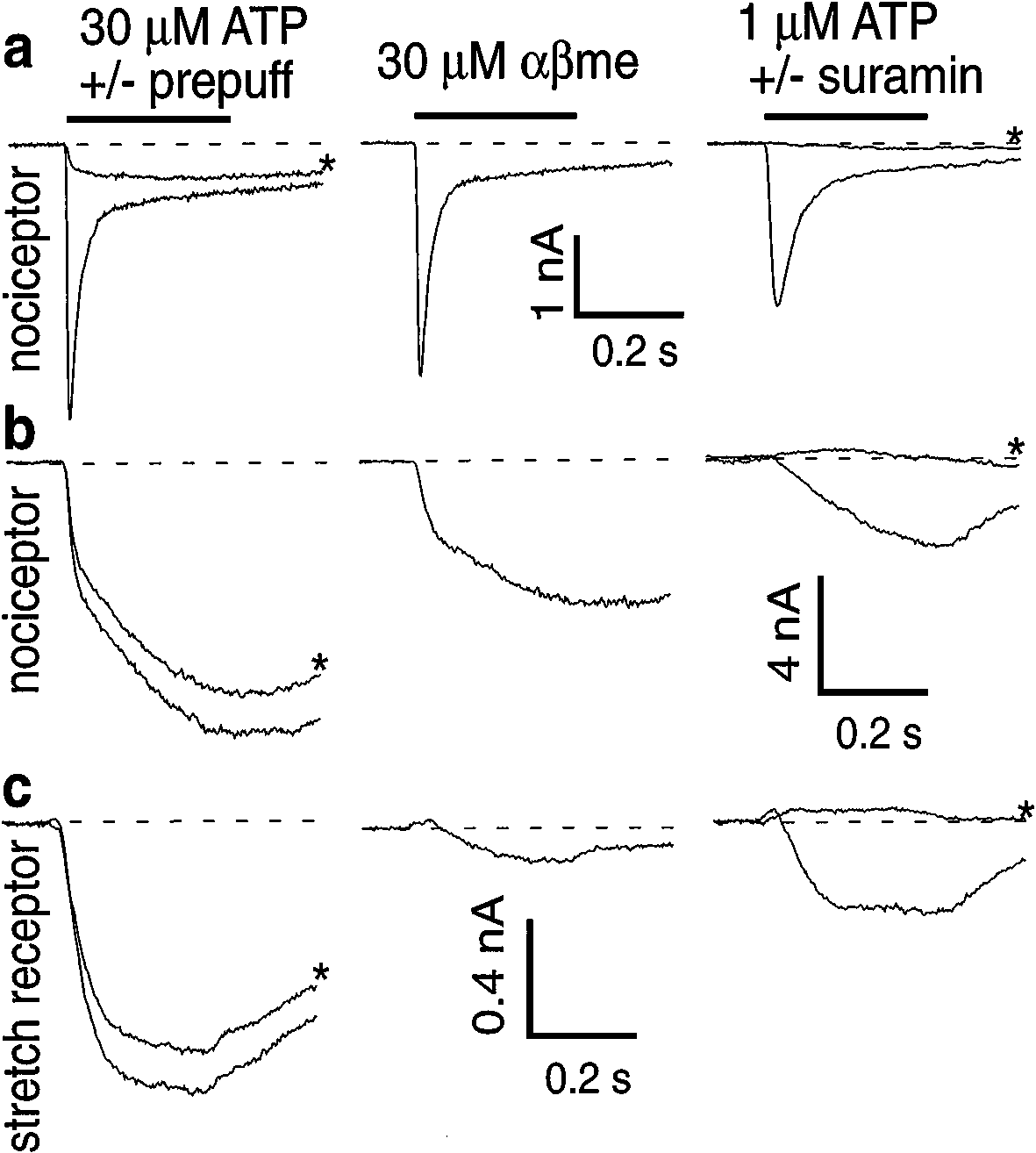 Nociceptors contain P2X3 receptors; stretch receptors do not. (a) Recordings from a
tooth pulp nociceptor enriched in transient type of ATP-gated current. Current is desensitized by anATP application 15 s prior to the stimulus (prepuff, left column); it is activated by α,β-methylene-ATP (middle column); it is blocked by suramin (right column). This pharmacology and the rapiddesensitization kinetics confirm the channel to be a P2X3 homomer. (b) The persistent ATP-gatedcurrent in nociceptors is activated by α,β-methylene-ATP and blocked by suramin, confirming itas a P2X2/P2X3 heteromer. (c) ATP-gated currents in stretch receptors are all persistent, but notactivated by α,β-methylene-ATP, unlike the persistent current in nociceptors. (From Reference 39.)
receptors at micromolar concentrations and the other P2X receptors at substan-tially higher concentrations (30). However, they are dirty drugs that inhibita variety of molecules. A more promising antagonist has recently appeared:Trinitrophenyl-ATP (ATP-TNP) selectively inhibits P2X1 and P2X3 recep-tors and heteromeric channels that contain one of these receptors as a subunit(42). A selective agonist, α,β-methylene-ATP, activates P2X1 and P2X3 re-ceptors (and some heteromeric channels containing these receptors) at far lower
concentrations than are needed for the other receptors (30). Distinguishing P2X1and P2X3 homomeric channels has been difficult because they both desensitize(albeit at noticeably different rates) and have similar pharmacology. Rae et al(43) argue that β,γ -methylene-L-ATP activates P2X1, but not P2X3, channels;this would indicate that the transient ATP-gated current in sensory neurons isentirely due to P2X3 receptors.
Nociceptors contain P2X3 receptors; stretch receptors do not. (a) Recordings from a
tooth pulp nociceptor enriched in transient type of ATP-gated current. Current is desensitized by anATP application 15 s prior to the stimulus (prepuff, left column); it is activated by α,β-methylene-ATP (middle column); it is blocked by suramin (right column). This pharmacology and the rapiddesensitization kinetics confirm the channel to be a P2X3 homomer. (b) The persistent ATP-gatedcurrent in nociceptors is activated by α,β-methylene-ATP and blocked by suramin, confirming itas a P2X2/P2X3 heteromer. (c) ATP-gated currents in stretch receptors are all persistent, but notactivated by α,β-methylene-ATP, unlike the persistent current in nociceptors. (From Reference 39.)
receptors at micromolar concentrations and the other P2X receptors at substan-tially higher concentrations (30). However, they are dirty drugs that inhibita variety of molecules. A more promising antagonist has recently appeared:Trinitrophenyl-ATP (ATP-TNP) selectively inhibits P2X1 and P2X3 recep-tors and heteromeric channels that contain one of these receptors as a subunit(42). A selective agonist, α,β-methylene-ATP, activates P2X1 and P2X3 re-ceptors (and some heteromeric channels containing these receptors) at far lower
concentrations than are needed for the other receptors (30). Distinguishing P2X1and P2X3 homomeric channels has been difficult because they both desensitize(albeit at noticeably different rates) and have similar pharmacology. Rae et al(43) argue that β,γ -methylene-L-ATP activates P2X1, but not P2X3, channels;this would indicate that the transient ATP-gated current in sensory neurons isentirely due to P2X3 receptors.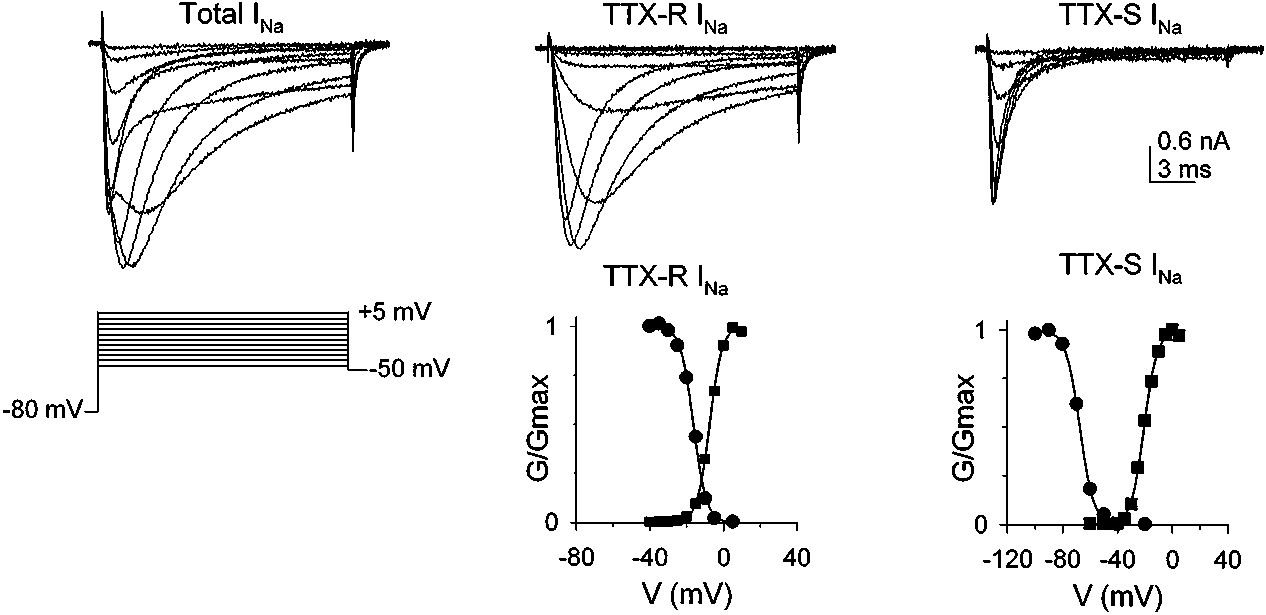 inactivation occurs more rapidly in TTX-resistant Na+ currents than in theTTX-sensitive currents present in uninjured neurons, it has been proposed thatneurons with a large proportion of TTX-resistant Na+ currents would be able tofire long trains of action potentials (88). The relative contribution of differentVGSCs to action potential generation in afferent terminals is unknown. How-ever, there is a growing body of evidence indicating that in normal tissue, TTX-resistant Na+ current is critically involved in the process (41, 92, 93) (Figure 2).
inactivation occurs more rapidly in TTX-resistant Na+ currents than in theTTX-sensitive currents present in uninjured neurons, it has been proposed thatneurons with a large proportion of TTX-resistant Na+ currents would be able tofire long trains of action potentials (88). The relative contribution of differentVGSCs to action potential generation in afferent terminals is unknown. How-ever, there is a growing body of evidence indicating that in normal tissue, TTX-resistant Na+ current is critically involved in the process (41, 92, 93) (Figure 2).