Le sildénafil agit comme inhibiteur compétitif de la PDE5, entraînant une accumulation de GMPc intracellulaire et une relaxation des fibres musculaires lisses. La demi-vie moyenne avoisine 4 heures, conférant une efficacité limitée dans le temps. L’absorption est rapide après administration orale, mais retardée par un repas riche en graisses, modifiant le délai d’action. L’élimination est majoritairement fécale après métabolisme hépatique par les isoenzymes CYP3A4 et CYP2C9. Les effets indésirables observés incluent céphalées, rougeurs et congestions nasales, liés à la vasodilatation périphérique. Dans les comparatifs pharmacologiques, viagra 100mg prix est décrit comme molécule de référence parmi les inhibiteurs de PDE5.
Pii: s0895-4356(00)00357-7
Journal of Clinical Epidemiology 54 (2001) 550–557
The randomized placebo-phase design for clinical trials
Brian Feldmana,*, Elaine Wangb, Andrew Willanc, John Paul Szalaid
aDivision of Rheumatology, The Hospital for Sick Children and the Department of Paediatrics and Public Health Sciences, University of Toronto, Toronto, Ontario, Canada
bDepartment of Paediatrics, The Hospital for Sick Children and the University of Toronto, Toronto, Ontario, Canada
cDepartment of Clinical Epidemiology and Biostatistics, McMaster University, Hamilton, Ontario, Canada
dDepartment of Research Design and Biostatistics, Sunnybrook and Women’s College Health Sciences Centre,and the University of Toronto, Toronto, Ontario, CanadaReceived 27 September 1999; received in revised form 2 October 2000; accepted 1 November 2000Abstract
Randomized controlled trials are the criterion standard method for evaluating the effectiveness of medical treatments. There are situ-
ations, however, where the possibility of being in the control group in a randomized controlled trial is unacceptable to potential subjectsor their physicians. This lack of acceptance is a reason for poor accrual. We developed and validated a new clinical trial design for survivaldata that may allay concerns about not receiving an investigational product and should be more acceptable. Called the randomized pla-cebo-phase design, this new design asks whether, on average, those subjects who begin active treatment sooner respond sooner than thosewho begin it later. Using Monte Carlo computer simulations, we demonstrated that the design is valid and may offer advantages over tra-ditional randomized controlled trials in some situations. The randomized placebo-phase design may be especially useful when highly po-tent therapies for rare diseases are tested or when accrual may be otherwise difficult.
2001 Elsevier Science Inc. All rights reserved. Keywords: Clinical trials; Randomization; Blinding; Therapy; Statistics; Efficacy
1. Introduction
rare that there are not enough subjects available for studythe power to detect a true treatment effect will be too lim-
Medicine has entered the evidence-based era, one in
ited to carry out a comparative trial. These two issues likely
which scientific evidence determines the best clinical prac-
interact: problems with the acceptability of a study make it
tice. This evidence comes from high-quality clinical studies,
harder to recruit subjects [1] which makes the recruitment
which in turn should form the basis for current treatments.
difficult when the condition being studied is uncommon [3].
However, providing studies of sufficient rigor to support
For example, a recent study [4] of the use of intravenous im-
treatment recommendations is difficult in many areas of
munoglobulin therapy for childhood arthritis failed because
medicine. Perceived problems with randomized controlled
of poor accrual. For relatively rare conditions, such as child-
trials (RCTs) have led, in some situations, to poor accept-
hood arthritis, there is a striking paucity of RCTs on which
ability by participants, physicians, and researchers [1].
The standard two-arm RCT is the criterion standard for the
Some patients, and their referring physicians, cannot ac-
evaluation of medical therapies [2] because the RCT is de-
cept treatments that are chosen by a process that is analogous
signed to limit bias, such as selection and measurement bias.
to the toss of a coin [6–8]. Investigators may also have prob-
In some situations, however, an RCT may be difficult to
lems with the acceptability of an RCT. If an investigator
carry out. For example, when a new therapy offers a poten-
knows, or has reason to believe, that one treatment arm is su-
tial cure for a previously untreatable and fatal disease, it
perior to another it is not ethical to enroll patients in an RCT.
may be unacceptable to have a control group that does not
Investigators are required to be in a state of equipoise [8].
receive the new therapy. In addition, when a disease is so
Even when the current slate of knowledge allows for reason-able equipoise in the wider medical community, treatment tri-als are usually conducted with the hope, perhaps even the ex-
* Corresponding author. The Hospital for Sick Children, 555 Univer-
pectation, that the experimental therapy will be better in some
sity Ave, Toronto ON M5B 1X8, Canada.
way than the traditional therapy or the placebo. Individual in-
Tel.: 416-813-5828; fax: 416-813-4989E-mail address:[email protected] (B. Feldman)
vestigators may not believe they have equipoise [9,10].
0895-4356/01/$ – see front matter 2001 Elsevier Science Inc. All rights reserved. PII: S 0 8 9 5 - 4 3 5 6 ( 0 0 ) 0 0 3 5 7 - 7
B. Feldman et al. / Journal of Clinical Epidemiology 54 (2001) 550–557
Investigators may use a variety of more powerful designs
the course of eight subjects, divided into two groups of four,
in order to study new therapies with fewer subjects. Cross-
who begin an effective experimental treatment at different
over trials and n-of-1 studies provide precise information
about the treatment response of individual subjects. Cross-
Selection bias might occur if those subjects who were
over trials may therefore be more statistically powerful than
more likely to respond sooner were assigned to start treat-
the parallel design RCT [11–13]. These designs, however,
ment sooner than those subjects likely to respond later. This
do not provide the same quality of information as the RCT,
is prevented in the RPPD by randomization of subjects to
and they can be used only when the treatment under study is
Many new therapies, for example monoclonal antibodies
directed against cell receptors (so-called biologics) andgene therapy, are aimed at producing a permanent responseor remission [14–17]. These treatments cannot be studiedwith crossover designs. Other study designs, such as therandomized responder [18] and randomized withdrawal de-sign [19], enroll subjects who have previously shown atreatment response in an open, single-arm phase. Two re-cent studies evaluating biologic treatments for childhood ar-thritis have used the randomized withdrawal design becauseit was felt by the investigators that a placebo controlledstudy would be perceived as being unacceptable by enroll-ing physicians and by patients [20,21]. This design, whileapparently acceptable, does not provide the same quality ofinformation as an RCT, and may require more subjects;only those that show an initial response are entered into therandomized portion of the study.
Because of the limitations of current research designs for
evaluating treatment efficacy in the context of rare diseaseslike childhood arthritis, a new study design was evaluated[22]. This design, called the randomized placebo-phase de-sign (RPPD), was developed to study remission-inducing(disease-modifying) therapies using survival endpoints. Allsubjects will receive active treatment is such a study design. This may lead to greater study acceptance and, in turn,greater recruitment. We describe the features of the RPPDand its usefulness for the study of rare disease, such aschildhood arthritis, by means of computer simulations. 2. Development of the randomized placebo-phase design
We reasoned that the most acceptable clinical trial design
would be one in which either the fewest subjects were as-signed to the placebo treatment or subjects were assigned tothe placebo treatment for only a short time. The RPPD fol-
Fig. 1. Features of the randomized placebo-phase design (RPPD) trial. (A)
lows the second strategy. It belongs to the class of designs
Overview of the RPPD. The vertical bars represent the start and finish
that includes the RCT in which the assignment of subjects is
dates of an RPPD clinical trial. The horizontal arrow indicates the direction
random and masked from subjects and assessors to reduce
of time. Each horizontal line represents one study subject. The triangles
bias. But, in an RPPD, unlike an RCT, all patients receive
signify the time when each subject starts the experimental therapy. Thesolid lines represent the time the subjects spend taking the experimental
the experimental therapy. It is the time from the enrollment
therapy. Subjects are followed until they respond (closed circle). (B) Rep-
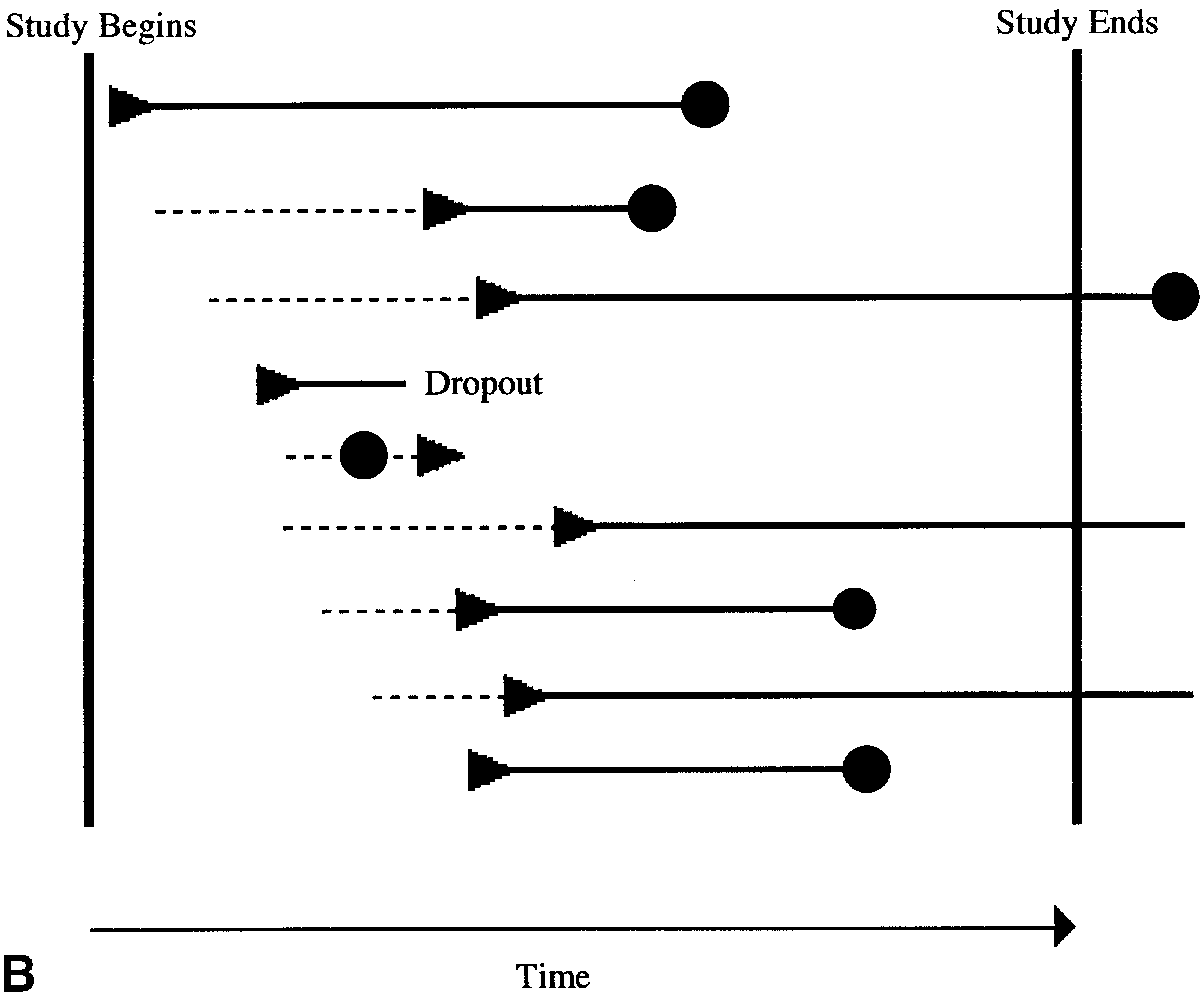
in the study to the commencement of the experimental treat-
resentation of an actual RPPD trial. In actual RPPD trials, subjects enter
ment that is randomized in the RPPD.
the trial at various times in the accrual phase. Subjects are randomly
The design of the RPPD assumes that a response occurs
assigned a period of time on placebo treatment (broken line). At the ran-
some time after an effective treatment is begun (during
domly determined time subjects blindly switch to active experimental ther-apy (triangles). Subjects who do not respond during the trial (third, sixth
which time the treatment effect takes place). Thus, if a ther-
and eighth subjects from top), and those who drop out (fourth subject from
apy is truly effective, those subjects who begin it sooner
top) are censored. Some subjects may respond spontaneously while still on
should, on average, respond sooner. Figure 1a demonstrates
B. Feldman et al. / Journal of Clinical Epidemiology 54 (2001) 550–557
The design shown in Figure 1a is susceptible to measure-
first set of simulations we studied a highly potent treatment,
ment bias; subjects and investigators, knowing which subjects
typical of the biologic treatments used in arthritis, that produces
started treatment sooner, may be more likely to think that there
a reliable and often relatively rapid response [21]. The second
is a treatment response sooner than if treatment was started late.
set of simulations studied a therapy of moderate potency, also
In the RPPD, this is limited by blinding subjects and investiga-
seen with biologic treatments. In the third set of simulations, we
tors. This blinding is accomplished by starting all subjects on
used a treatment of intermediate potency. An example of this
an identically appearing and tasting placebo that is switched to
sort of treatment would be methotrexate for the treatment of
active treatment at the randomly determined time. Subjects and
childhood arthritis [24]. Finally, we studied a low potency treat-
investigators are unaware of when the switch takes place. Some
ment, to which the response is highly variable and may take a
subjects may be assigned a placebo phase length of 0: they start
long time, similar to gold in the treatment of arthritis [25].
the study taking active treatment. The random duration of theplacebo phase gives the RPPD its name.
The statistical power of the RPPD would be maximized
Subjects were enrolled in RPPD trials as described.
if the placebo phase length was limited to two values
There was a fixed accrual period. Each subject was then
(namely, 0 and the maximum allowed length). However, to
randomly assigned a placebo duration, after which therapy
preserve blinding there may be situations when subjects
with the experimental agent was begun. Subjects were then
should be randomly assigned to a placebo phase that might
followed until a defined response was achieved, they
dropped out, or until the trial ended.
During such a clinical trial (represented by Figure 1b)
We assumed a survival (time-to-event) endpoint. We
subjects would be recruited at various times after the trial
have not evaluated the RPPD for continuous outcomes. It
start date. The time taken to respond to active treatment
was assumed that the time until response followed an expo-
would not be fixed, but would vary from one subject to the
nential distribution. The exponential distribution is com-
next. Some subjects would be censored; that is, they would
monly used to model time-to-event data [26] including time
drop out before they respond or finish the trial without re-
to treatment responses and time to developing adverse ef-
sponding. Drop out might occur completely at random, or
fects from medications. The exponential distribution of the
might be related in some way to the treatment given, or to
time to response assumes that the probability of a subject re-
the observation process (see discussion below). In the
sponding at any time (assuming he or she has not yet re-
RPPD, a Cox proportional hazards regression [23] is used to
sponded) remains constant for the length of the trial. It is not
determine the statistical significance of the observed treat-
known how often this assumption holds true for new experi-
ment effect. The dependent variable is the time from the en-
mental treatments. However, there is some evidence that
try into the study to the time of a response to the treatment.
treatment responses in pediatric rheumatic diseases can be
The independent variable is the time from the entry into the
adequately characterized by the exponential distribution
study to the time of starting experimental therapy, in other
[22]. For example, a recent study of a biologic agent used in
words, the length of the placebo phase. The length of the
the treatment of childhood arthritis produced responses that
placebo phase will predict the overall time to a response, if
followed an exponential distribution [21, M. Lange (Immu-
3. Characterization of the RPPD
The simulation program was written in the SAS pro-
gramming language (version 6.12). All simulations were
done on a Digital DEC 5000 minicomputer. Simulation re-sults were analyzed with DataDesk 6.1 [27].
To determine how efficient, how powerful, the RPPD de-
sign is, we compared RPPD trials with standard, parallel-
3.2.4. Number of iterations and sample size
arm RCTs, using a Monte Carlo computer simulation.
Each simulation run consisted of 500 iterations, or 500
Under the conditions of our assumptions, we hypothe-
sized that the RPPD would be less powerful than the RCT if
The sample size for each RPPD clinical trial was varied
full recruitment into the RCT occurred. Because our simula-
tions assumed that the treatment–response times would beexponentially distributed (see below), we hypothesized that
3.2.5. Accrual phase and length of trial
the RPPD would approach the RCT in power for the study
In a typical clinical trial, subjects are accrued over a
fixed period early in the study. In these simulations, the ac-crual phase was kept constant at 90 days and the total trial
We simulated four types of therapies that might apply to the
In an RPPD trial, subjects are randomly assigned to a
treatment of rare diseases, such as childhood arthritis. In the
varying duration of placebo therapy before starting active
B. Feldman et al. / Journal of Clinical Epidemiology 54 (2001) 550–557
therapy. For this study, individual subjects were randomly
assigned a placebo phase length of 0 or 60 days with equal
For the baseline simulations it was assumed that no drop-
probability. To characterize the loss of statistical power as-
outs occurred. We then repeated the simulations under the
sociated with allowing the placebo phase to take on a
assumption that an average of 10% and 20% of the subjects
greater number of values, we repeated the simulations and
dropped out before a response was seen. Missing data in the
randomly assigned subjects to placebo phase lengths of 0 to
form of dropout can be classified as completely random
dropout, random dropout or informative dropout [28]. Inour simulations, subjects dropped out in a completely ran-
3.2.7. Median time to a response, untreated
dom manner. In an actual clinical trial, the dropout process
In some diseases, such as arthritis, subjects may have a
may in fact be informative necessitating a more complex
spontaneous response, without specific therapy. This phe-
nomenon was accounted for in the assumptions. We used aconservative value of a median time to spontaneous re-
sponse of 300 days (a baseline daily hazard of 0.0023). Un-
Each simulated trial was analyzed with the Cox propor-
der this assumption about 20% of the subjects will have a
tional hazards regression [23]. The dependent variable was
spontaneous response within 3 months. This is similar to the
the time from entry into the trial until the time when a re-
placebo response seen in many arthritis clinical trials.
sponse occurred. The independent variable was the lengthof the placebo phase. Subjects who did not respond by the
3.2.8. Median time to a response, treated
The median response for the highly potent treatment was
The Cox regression yields a likelihood-ratio statistic,
assumed to be 7 days (a treated daily hazard of 0.099) and
which, when the treatment has no effect, has a chi-square
for the moderately potent treatment 14 days (hazard of
distribution with 1 degree of freedom. A value greater than
0.050). The intermediate potency treatment was assumed to
3.841 is critical for an alpha-error (a false-positive) proba-
bring about a response at a median of 42 days (treated daily
bility of 0.05. Therefore, a trial was considered positive if
hazard of 0.017) while the low potency treatment had a me-
those randomized to be treated sooner were observed to re-
dian response time of 150 days (hazard of 0.0046).
spond sooner, and the resulting likelihood-ratio statistic was
Fig. 2. The power curves for the RPPD. The y-axis represents the power (1-B) expressed as a percentage. The x-axis represents total sample size on a logarith-mic scale. Each of the four curves represents simulations of treatments of different potency as described in Methods. B. Feldman et al. / Journal of Clinical Epidemiology 54 (2001) 550–557
greater than or equal to 3.841. The validity of this cut-off
was confirmed in a previous simulation study [22] by the
calculation of the fraction of false-positive trials when an
experimental therapy of no effect was simulated.
Power (1 Ϫ beta) was calculated as the fraction of trials
(out of 500) in each simulation run that was positive. For
each simulated therapy, the results of simulation runs were
plotted against sample size to develop power curves.
The power of RPPD simulated studies was compared
with the power of RCTs. RCT scenarios used the same as-sumptions and same total sample size as the RPPD simula-tions, but the subjects were randomized into treatment andplacebo arms with equal probability. The power of the
intermediate potency treatment is studied, and about 240 sub-
RCTs was determined with standard calculations (the time
jects to reach a power of 0.80 for the low potency therapy.
to a response in a parallel-group two-arm placebo-con-
Table 1 displays the required sample size to reach a
trolled trial analyzed with the log-rank test) [29].
power of 0.80 and 0.90 for each simulated therapy and forthe corresponding parallel arms RCT. As hypothesized, theRCT is more powerful, a difference that becomes moremarked with less potent treatments. 4. Results
The effect of a 10% and a 20% dropout rate is displayed
The power curves for the RPPD are shown in Figure 2.
in Figure 3 for the highly potent treatment. Under our as-
For a highly potent therapy the RPPD reaches a statistical
sumption of completely random dropout, the RPPD is quite
power of Ͼ0.80 with seven subjects. The statistical power
drops off with less potent therapies. For example, the RPPD
Figure 4 shows the corresponding power curves for the
requires about 31 subjects to reach a power of 0.80 when the
RPPD when the placebo phase is allowed to vary between 0
Fig. 3. The power curves for the RPPD studying a highly potent therapy (see Methods). The y-axis represents power expressed as a percentage. The x-axisrepresents total sample size. The solid line represents the baseline case. The other lines represent simulations in which 10% and 20% of subjects drop out onan average. B. Feldman et al. / Journal of Clinical Epidemiology 54 (2001) 550–557
and 60 days. There is a marked loss of statistical power
variability from a small amount of experimental, or random,
more apparent for treatments of lower potency.
In contrast, highly potent treatments with a very rapid
onset have much less variance in response times. Thiswould suggest that the RPPD might be more useful in the
5. Discussion
study of treatments such as analgesics, antibiotics, and bio-
We found that the RPPD clinical trial was able to yield
logics, which exert their effects rapidly. Even with highly
positive results when experimental therapies of varying effi-
potent treatments an RCT would require a smaller sample
cacy were studied. These results validate the concept of a
size than an RPPD; the decision to implement an RPPD trial
treatment trial in which those subjects who are treated
would depend on the perceived acceptability of a standard
sooner are expected, on average, to respond sooner than
We believe that in special circumstances the RPPD
In addition, our simulations show that the power of the
should be more acceptable to patients, physicians, and in-
RPPD, while adequate for highly potent therapies, may not
vestigators. In this design, no subjects are required to take a
be adequate for therapies that have a more variable result
placebo for more than a relatively short time. All subjects,
and a longer average time to response. These results were
therefore, know that they will receive the experimental
predictable, given the assumptions underlying the simula-
treatment; furthermore, physicians’ ethical misgivings
tions. We assumed that the response to treatment would fol-
about RCTs, whether theoretically correct or not, are al-
low an exponential distribution (i.e., would be characterized
layed. This improvement in the acceptability of the study
by a constant conditional probability of a response). The
design should mean greater accrual. At this point, the
variance of response times under the exponential distribu-
greater acceptability of this new design, while reasonable, is
tion is the square of the mean response time [26]. Therefore,
speculative. Only practical application will be able to deter-
the variance in response times was much larger for those
mine whether accrual is truly enhanced.
treatments that tended to take a long time to achieve a re-
The RPPD might be considered early in the study of new
sponse. In our studies of low and intermediate potency ther-
treatments. Currently, in new treatment development, the
apies, we were trying to predict a large amount of response
earliest studies are often open-label case-series. In the
Fig. 4. The power curves for the RPPD. The y-axis represents the power (1-B) expressed as a percentage. The x-axis represents total sample size on a logarith-mic scale. Each of the four curves represents simulations of treatments of different potency as described in Methods. In this set of simulations the randomlyassigned placebo phase was allowed to vary anywhere from 0 to 60 days. B. Feldman et al. / Journal of Clinical Epidemiology 54 (2001) 550–557
RPPD design the assignment to the different lengths of the
a useful method for studying new therapies, especially as an
placebo phase is random to limit bias. Likewise, measure-
alternative to open, uncontrolled trials.
ments of the response to therapy are blind to the knowledgeof the length of the placebo phase. In these ways, the RPPD
Acknowledgments
is more rigorous than, and should be preferable to, the case-series design.
We gratefully acknowledge Dr. Colin MacArthur for his
These advantages are offset by a decrease in statistical
critical review of our manuscript. This article was prepared
power and, more importantly, by limitations in the ability to
with the assistance of Editorial Services, The Hospital for
draw inferences about the size of the treatment effect.
Sick Children, Toronto, Ontario, Canada.
In our simulations, the RPPD was always less powerful
This work was presented in part at the American College
than the two-arm placebo-controlled RCT with the same to-
of Rheumatology Annual Meeting, San Francisco, 1995.
tal sample size; this difference was greater with treatmentsof low potency. Likely, the advantages of the RPPD,namely, potentially greater acceptance and higher accrual,
References
would be apparent only when used to study highly potent
[1] Gotay CC. Accrual to cancer clinical trials: directions from the re-
therapies. However, in situations in which it is completely
search literature. Soc Sci Med 1991;33:569–77.
unacceptable to have a two-arm controlled trial and in
[2] Sackett D, Haynes R, Tugwell P. Clinical Epidemiology—A Basic
which many subjects are available, the RPPD may be pref-
Science for Clinical Medicine. Boston, MA: Little, Brown, 1985.
[3] Feldman BM, Giannini EH. Where’s the evidence? Putting clinical
erable to an open case-series design, even for low potency
science into pediatric rheumatology. J Rheumatol 1996;23:1502–4.
treatments. Examples of such situations might be the early
[4] Silverman ED, Cawkwell GD, Lovell DJ, Laxer RM, Lehman TJ,
testing of a new anti-tumor agent directed toward a cur-
Passo MH, Zemel LS, Giannini EH. Intravenous immunoglobulin in
rently untreatable cancer or the testing of a potentially cura-
the treatment of systemic juvenile rheumatoid arthritis: a randomized
placebo controlled trial. J Rheumatol 1994;21:2353–8.
[5] Rosenberg AM: Treatment of JRA: approach to patients who fail
The RPPD does not permit the direct comparison of the
standard therapy. J Rheumatol 1996;23:1652–6.
effects of a placebo with those of the experimental therapy.
[6] Schafer A. The ethics of the randomized clinical trial. N Engl J Med
Investigators will, however, be able to estimate the average
length of time until a treatment response occurs and the pro-
[7] Kotwall CA, Mahoney LJ, Myers RE, DeCoste L. Reasons for non-
portion of subjects who respond at any point in time. In ad-
entry in randomized clinical trials for breast cancer: a single institu-tional study. J Surg Oncol 1992;50:125–9.
dition, they will be able to determine the probability that the
[8] Freedman B. Equipoise and the ethics of clinical research. N Engl J
treatment has an effect beyond chance. For this reason, the
RPPD would probably be best used early in the develop-
[9] Schulz KF. Unbiased research and the human spirit: the challenges of
ment of a new treatment when accrual to an RCT would be
randomized controlled trials. Can Med Assoc J 1995;153:783–6.
difficult or in place of open, uncontrolled studies (see
[10] Klein MC, Kaczorowski J, Robbins JM, Gauthier RJ, Jorgensen SH,
Joshi AK. Physicians’ beliefs and behaviour during a randomized
above). For situations in which a definitive RCT would be
controlled trial of episiotomy: consequences for women in their care.
unacceptable to impossible, the RPPD could be considered
for later stages of the development of treatment.
[11] Hills M, Armitage P. The two-period cross-over clinical trial. Br J
Our simulations provide only a limited understanding of
the design features of the RPPD. For example, we used a
[12] Johannessen T, Fosstvedt D, Petersen H. Combined single subject tri-
als. Scand J Primary Health Care 1991;9:23–7.
number of simplifying assumptions that may not apply in
[13] Jaeschke R, Adachi J, Guyatt G, Keller J, Wong B. Clinical useful-
actual studies. We assumed an exponential distribution of
ness of amitriptyline in fibromyalgia: the results of 23 N-of-1 ran-
response times. Under other assumptions, the power of the
domized controlled trials. J Rheumatol 1991;18:447–51.
RPPD might be much larger or smaller. In actual studies,
[14] Otani K, Nita I, Macaulay W, Georgescu HI, Robbins PD, Evans CH.
the distribution of the time to response may not be known
Suppression of antigen-induced arthritis in rabbits by ex vivo genetherapy. J Immunol 1996;156:3558–62.
and may be complex. There is little evidence to guide the
[15] Bandara G, Mueller GM, Galea-Lauri J, Tindal MH, Georgescu HI,
choice for a treatment response distribution for simulation
Suchanek MK, Hung GL, Glorioso JC, Robbins PD, Evans CH. In-
studies. We chose the exponential distribution for our simu-
traarticular expression of biologically active interleukin 1-receptor-
lations because of its familiarity; in addition we have data
antagonist protein by ex vivo gene transfer. Proc Natl Acad Sci 1993;
that at least some treatment responses follow an exponential
[16] Kavanaugh AF, Davis LS, Nichols LA, Norris SH, Rothlein R,
distribution [21,22]. Further work needs to be done in this
Scharschmidt LA, Lipsky PE. Treatment of refractory rheumatoid ar-
thritis with a monoclonal antibody to intercellular adhesion molecule
In summary, the RPPD offers a new study tool for spe-
cial circumstances where a standard RCT would be difficult
[17] Matteson EL, Yocum DE, St Clair EW, Achkar AA, Thakor MS, Ja-
or impossible. The RPPD is a potentially more acceptable
cobs MR, Hayes AE, Heitman CK, Johnston JM. Treatment of activerefractory rheumatoid arthritis with humanized monoclonal antibody
clinical trial design for survival endpoints. It permits valid
Campath-1H administered by daily subcutaneous injection. Arthritis
conclusions about treatment efficacy and has adequate sta-
tistical power for highly potent therapies. The RPPD may be
[18] Hallstrom AP, Verter J, Friedman L. Randomizing responders. Car-
B. Feldman et al. / Journal of Clinical Epidemiology 54 (2001) 550–557
diac Arrhythmia Suppression Trial (CAST) investigators. Control
[23] Cox D. Regression models and life-tables. J Royal Stat Soc 1972; Se-
[19] The Canadian Hydroxychloroquine Study Group. A randomized
[24] Giannini EH, Brewer EJ, Kuzmina N, Shaikov A, Maximov A,
study of the effect of withdrawing hydroxychloroquine sulfate in sys-
Vorontsov I, Fink CW, Newman AJ, Cassidy JT, Zemel LS. Metho-
temic lupus erythematosus. N Engl J Med 1991;324:150–4.
trexate in resistant juvenile rheumatoid arthritis: results of the USA–
[20] Giannini EH, Lovell DJ, Silverman ED, Sundel RP, Tague BL, Ru-
USSR double-blind, placebo-controlled trial. N Engl J Med 1992;
perto N. Intravenous immunoglobulin in the treatment of polyarticu-
lar juvenile rheumatoid arthritis: a phase I/II study. Pediatric Rheu-
[25] Wolfe F, Hawley DJ, Cathey MA. Measurement of gold treatment ef-
matology Collaborative Study Group. J Rheumatol 1996;23:919–24.
fect in clinical practice: evidence for effectiveness of intramuscular
[21] Lovell DJ, Giannini EH, Reiff A, Cawkwell GD, Silverman ED, Noc-
gold therapy. J Rheumatol 1993;20:797–801.
ton JJ, Stein LD, Gedalia A, Ilowite NT, Wallace CA, Whitmore H,
[26] Lee E. Statistical methods for survival data analysis. Belmont, CA:
Finck BK. Etanercept in children with polyarticular juvenile rheuma-
toid arthritis. Pediatric Rheumatology Collaborative Study Group. N
[27] Velleman P. Data desk Version 4.1. Ithaca, NY: Data Description, 1992.
[28] Diggle P, Kenward MG. Informative dropout in longitudinal data
[22] Feldman BM. Evaluation of a new single-arm randomized placebo
analyis. Appl Statist 1994;43:49–93.
phase design clinical trial using Monte Carlo computer simulation
[29] Dupont WD, Plummer WD. Power and sample size calculations: a re-
[MSc thesis]. Toronto, Ontario: University of Toronto, 1995.
view and computer program. Control Clin Trials 1990;11:116–28.
EFNS TASK FORCE ARTICLE Linee Guida EFNS per il trattamento della cefalea a grappolo e delle altre cefalee autonomico-trigeminali A. Maya, M. Leoneb, J. Áfrac, M. Linded, P. S. Sándore, S. Eversf and P. J. Goadsby aDepartment of Systems Neuroscience, University of Hamburg, Hamburg, Germany; bIstituto Neurologico Carlo Besta, Milan, Italy; cNational Institute of Neurosurgery, Budape
Conduct of seminars, discussions, round-tables and workshops devoted to the boardaspects of telecommunications policy, organisation, performance, technology re-search, consumer protection, telecom laws, etc., Involving providers and consumersof service, economists, intellectuals and policy makers; ICTs AND SOCIETY Involving consumer associations and providers of service in discussions; Editor

 Journal of Clinical Epidemiology 54 (2001) 550–557
The randomized placebo-phase design for clinical trials
Brian Feldmana,*, Elaine Wangb, Andrew Willanc, John Paul Szalaid
aDivision of Rheumatology, The Hospital for Sick Children and the Department of Paediatrics and Public Health Sciences,
University of Toronto, Toronto, Ontario, Canada
bDepartment of Paediatrics, The Hospital for Sick Children and the University of Toronto, Toronto, Ontario, Canada
cDepartment of Clinical Epidemiology and Biostatistics, McMaster University, Hamilton, Ontario, Canada
dDepartment of Research Design and Biostatistics, Sunnybrook and Women’s College Health Sciences Centre,
and the University of Toronto, Toronto, Ontario, Canada
Received 27 September 1999; received in revised form 2 October 2000; accepted 1 November 2000
Abstract
Journal of Clinical Epidemiology 54 (2001) 550–557
The randomized placebo-phase design for clinical trials
Brian Feldmana,*, Elaine Wangb, Andrew Willanc, John Paul Szalaid
aDivision of Rheumatology, The Hospital for Sick Children and the Department of Paediatrics and Public Health Sciences,
University of Toronto, Toronto, Ontario, Canada
bDepartment of Paediatrics, The Hospital for Sick Children and the University of Toronto, Toronto, Ontario, Canada
cDepartment of Clinical Epidemiology and Biostatistics, McMaster University, Hamilton, Ontario, Canada
dDepartment of Research Design and Biostatistics, Sunnybrook and Women’s College Health Sciences Centre,
and the University of Toronto, Toronto, Ontario, Canada
Received 27 September 1999; received in revised form 2 October 2000; accepted 1 November 2000
Abstract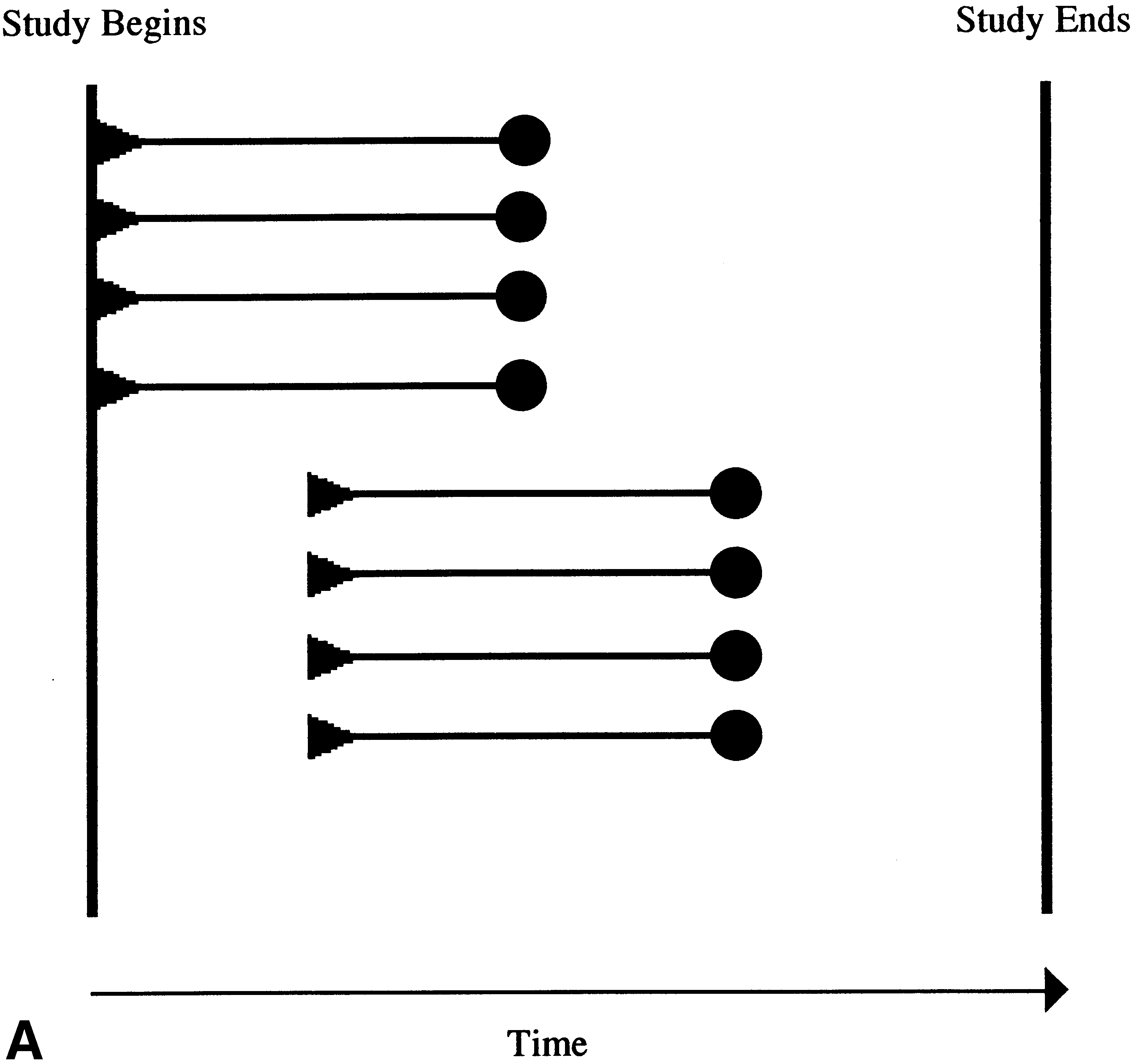
 B. Feldman et al. / Journal of Clinical Epidemiology 54 (2001) 550–557
Investigators may use a variety of more powerful designs
the course of eight subjects, divided into two groups of four,
in order to study new therapies with fewer subjects. Cross-
who begin an effective experimental treatment at different
over trials and n-of-1 studies provide precise information
about the treatment response of individual subjects. Cross-
Selection bias might occur if those subjects who were
over trials may therefore be more statistically powerful than
more likely to respond sooner were assigned to start treat-
the parallel design RCT [11–13]. These designs, however,
ment sooner than those subjects likely to respond later. This
do not provide the same quality of information as the RCT,
is prevented in the RPPD by randomization of subjects to
and they can be used only when the treatment under study is
Many new therapies, for example monoclonal antibodies
directed against cell receptors (so-called biologics) andgene therapy, are aimed at producing a permanent responseor remission [14–17]. These treatments cannot be studiedwith crossover designs. Other study designs, such as therandomized responder [18] and randomized withdrawal de-sign [19], enroll subjects who have previously shown atreatment response in an open, single-arm phase. Two re-cent studies evaluating biologic treatments for childhood ar-thritis have used the randomized withdrawal design becauseit was felt by the investigators that a placebo controlledstudy would be perceived as being unacceptable by enroll-ing physicians and by patients [20,21]. This design, whileapparently acceptable, does not provide the same quality ofinformation as an RCT, and may require more subjects;only those that show an initial response are entered into therandomized portion of the study.
B. Feldman et al. / Journal of Clinical Epidemiology 54 (2001) 550–557
Investigators may use a variety of more powerful designs
the course of eight subjects, divided into two groups of four,
in order to study new therapies with fewer subjects. Cross-
who begin an effective experimental treatment at different
over trials and n-of-1 studies provide precise information
about the treatment response of individual subjects. Cross-
Selection bias might occur if those subjects who were
over trials may therefore be more statistically powerful than
more likely to respond sooner were assigned to start treat-
the parallel design RCT [11–13]. These designs, however,
ment sooner than those subjects likely to respond later. This
do not provide the same quality of information as the RCT,
is prevented in the RPPD by randomization of subjects to
and they can be used only when the treatment under study is
Many new therapies, for example monoclonal antibodies
directed against cell receptors (so-called biologics) andgene therapy, are aimed at producing a permanent responseor remission [14–17]. These treatments cannot be studiedwith crossover designs. Other study designs, such as therandomized responder [18] and randomized withdrawal de-sign [19], enroll subjects who have previously shown atreatment response in an open, single-arm phase. Two re-cent studies evaluating biologic treatments for childhood ar-thritis have used the randomized withdrawal design becauseit was felt by the investigators that a placebo controlledstudy would be perceived as being unacceptable by enroll-ing physicians and by patients [20,21]. This design, whileapparently acceptable, does not provide the same quality ofinformation as an RCT, and may require more subjects;only those that show an initial response are entered into therandomized portion of the study.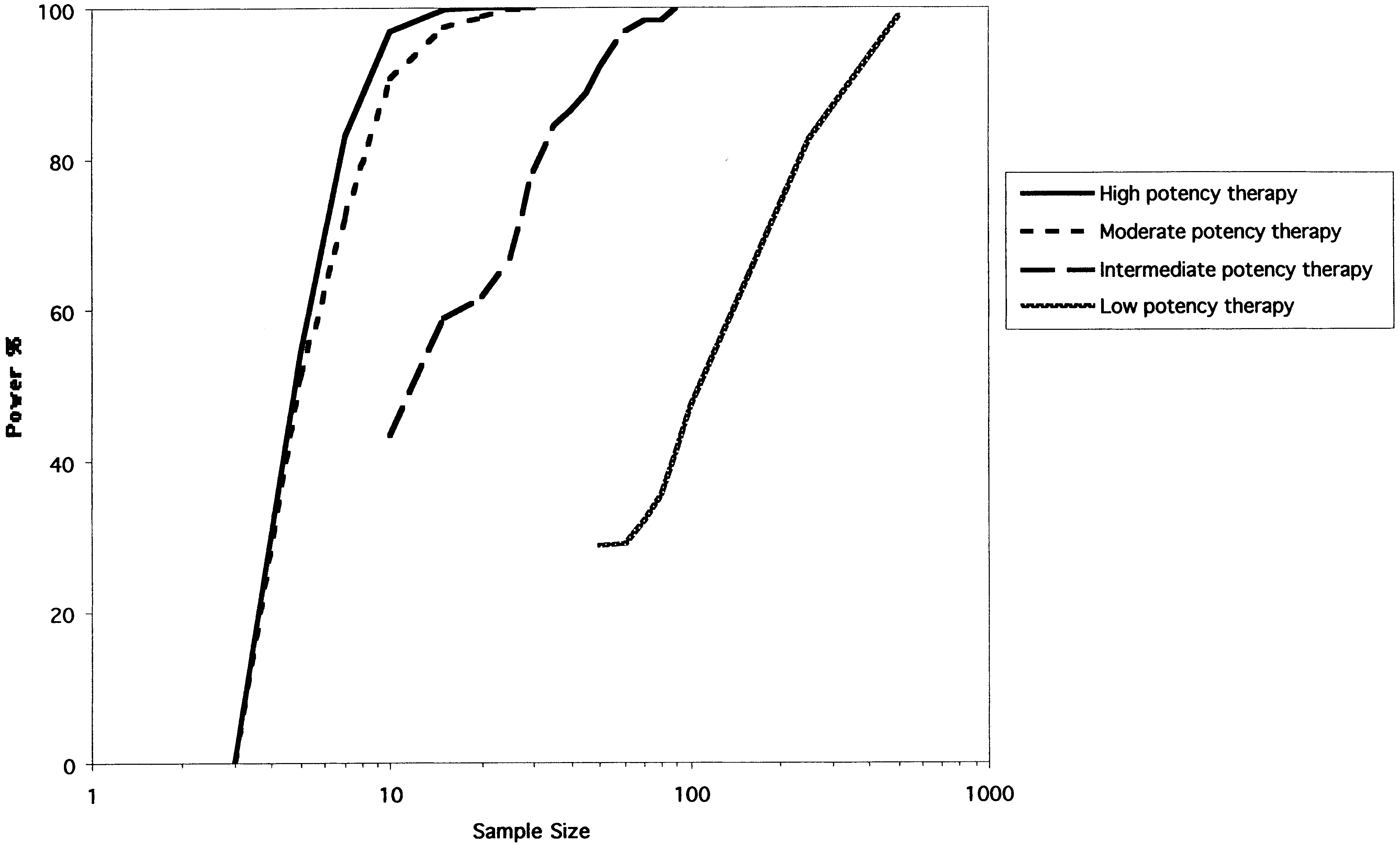 B. Feldman et al. / Journal of Clinical Epidemiology 54 (2001) 550–557
therapy. For this study, individual subjects were randomly
assigned a placebo phase length of 0 or 60 days with equal
For the baseline simulations it was assumed that no drop-
probability. To characterize the loss of statistical power as-
outs occurred. We then repeated the simulations under the
sociated with allowing the placebo phase to take on a
assumption that an average of 10% and 20% of the subjects
greater number of values, we repeated the simulations and
dropped out before a response was seen. Missing data in the
randomly assigned subjects to placebo phase lengths of 0 to
form of dropout can be classified as completely random
dropout, random dropout or informative dropout [28]. Inour simulations, subjects dropped out in a completely ran-
3.2.7. Median time to a response, untreated
dom manner. In an actual clinical trial, the dropout process
In some diseases, such as arthritis, subjects may have a
may in fact be informative necessitating a more complex
spontaneous response, without specific therapy. This phe-
nomenon was accounted for in the assumptions. We used aconservative value of a median time to spontaneous re-
sponse of 300 days (a baseline daily hazard of 0.0023). Un-
Each simulated trial was analyzed with the Cox propor-
der this assumption about 20% of the subjects will have a
tional hazards regression [23]. The dependent variable was
spontaneous response within 3 months. This is similar to the
the time from entry into the trial until the time when a re-
placebo response seen in many arthritis clinical trials.
B. Feldman et al. / Journal of Clinical Epidemiology 54 (2001) 550–557
therapy. For this study, individual subjects were randomly
assigned a placebo phase length of 0 or 60 days with equal
For the baseline simulations it was assumed that no drop-
probability. To characterize the loss of statistical power as-
outs occurred. We then repeated the simulations under the
sociated with allowing the placebo phase to take on a
assumption that an average of 10% and 20% of the subjects
greater number of values, we repeated the simulations and
dropped out before a response was seen. Missing data in the
randomly assigned subjects to placebo phase lengths of 0 to
form of dropout can be classified as completely random
dropout, random dropout or informative dropout [28]. Inour simulations, subjects dropped out in a completely ran-
3.2.7. Median time to a response, untreated
dom manner. In an actual clinical trial, the dropout process
In some diseases, such as arthritis, subjects may have a
may in fact be informative necessitating a more complex
spontaneous response, without specific therapy. This phe-
nomenon was accounted for in the assumptions. We used aconservative value of a median time to spontaneous re-
sponse of 300 days (a baseline daily hazard of 0.0023). Un-
Each simulated trial was analyzed with the Cox propor-
der this assumption about 20% of the subjects will have a
tional hazards regression [23]. The dependent variable was
spontaneous response within 3 months. This is similar to the
the time from entry into the trial until the time when a re-
placebo response seen in many arthritis clinical trials.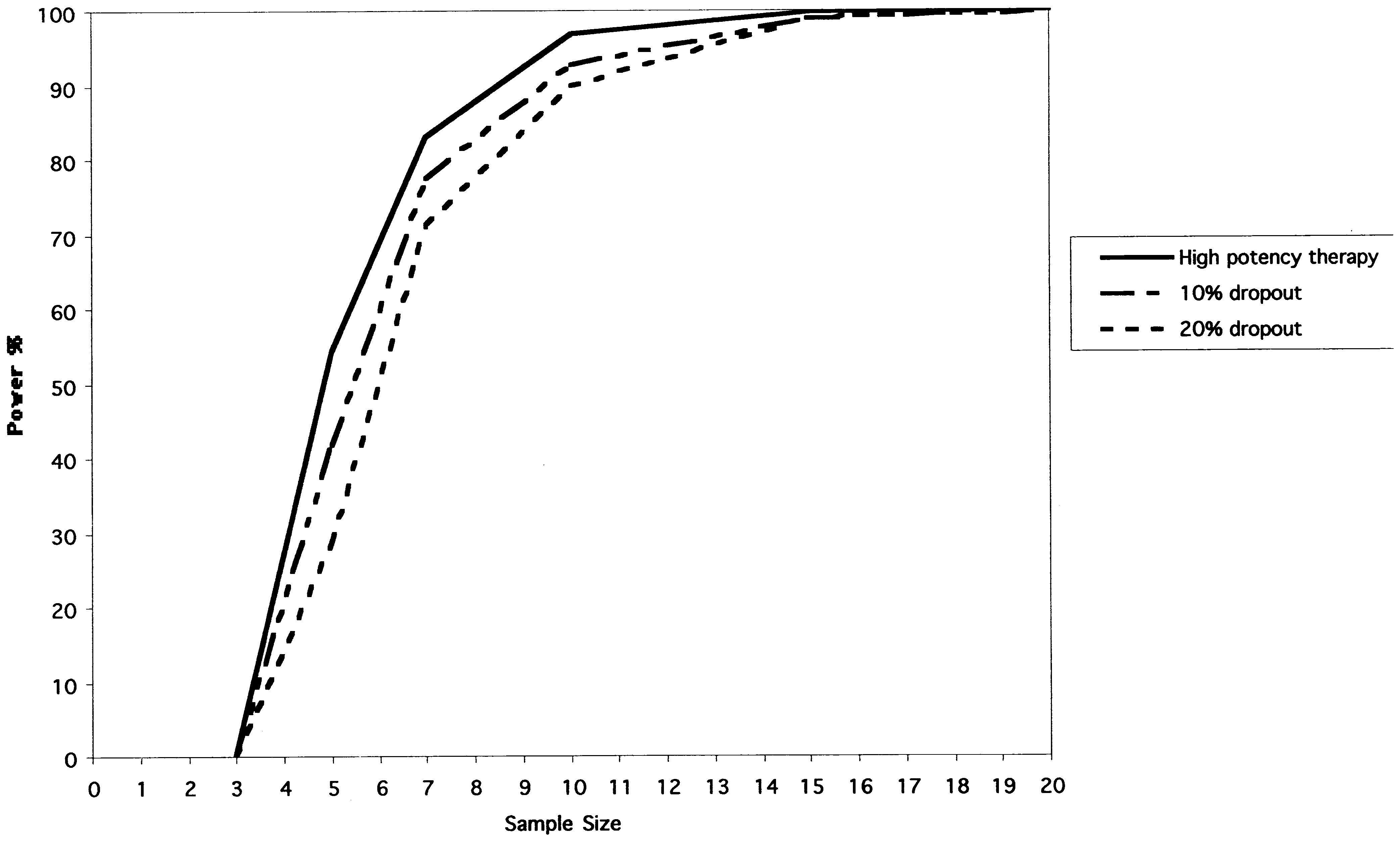 B. Feldman et al. / Journal of Clinical Epidemiology 54 (2001) 550–557
greater than or equal to 3.841. The validity of this cut-off
was confirmed in a previous simulation study [22] by the
calculation of the fraction of false-positive trials when an
experimental therapy of no effect was simulated.
B. Feldman et al. / Journal of Clinical Epidemiology 54 (2001) 550–557
greater than or equal to 3.841. The validity of this cut-off
was confirmed in a previous simulation study [22] by the
calculation of the fraction of false-positive trials when an
experimental therapy of no effect was simulated.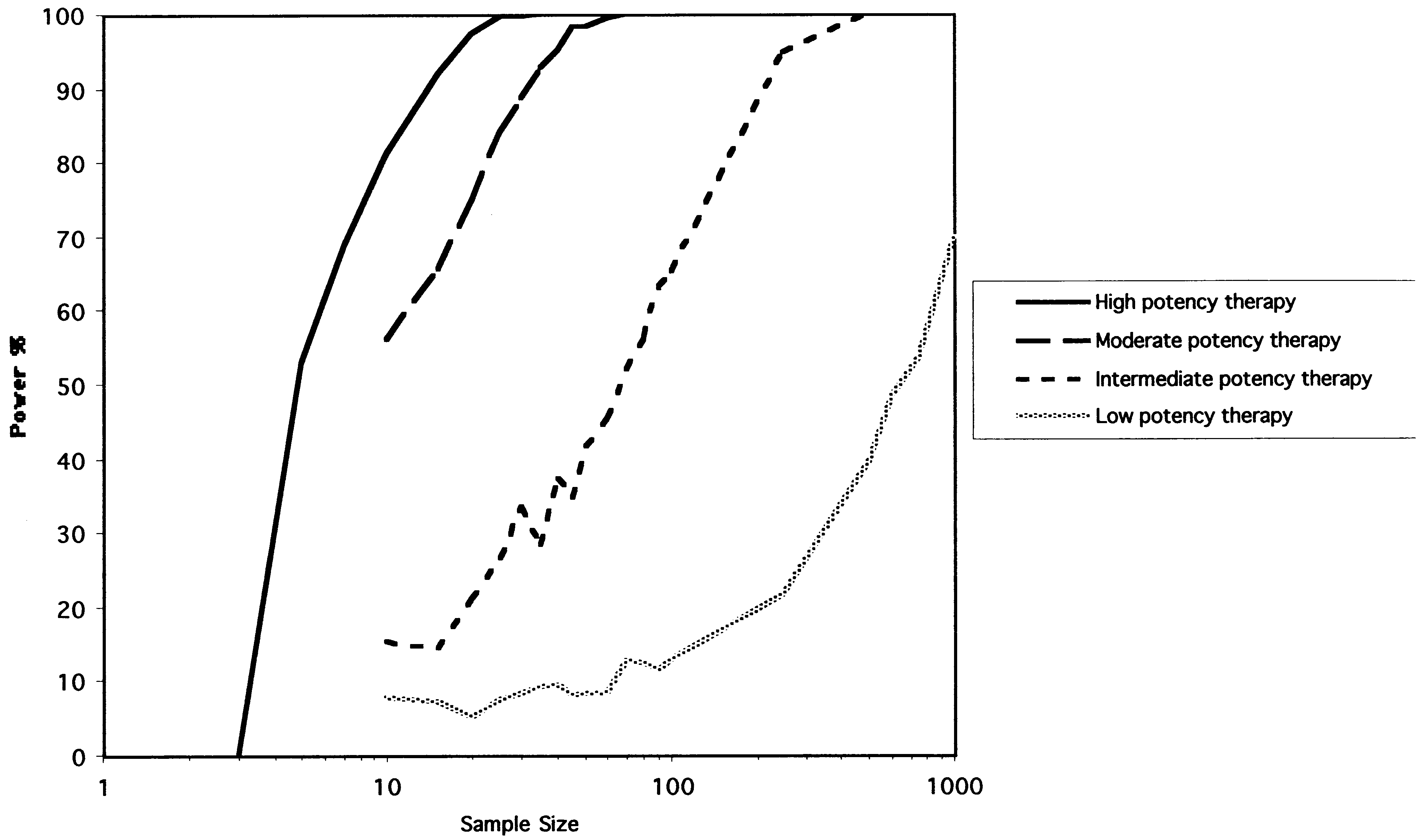 B. Feldman et al. / Journal of Clinical Epidemiology 54 (2001) 550–557
and 60 days. There is a marked loss of statistical power
variability from a small amount of experimental, or random,
more apparent for treatments of lower potency.
B. Feldman et al. / Journal of Clinical Epidemiology 54 (2001) 550–557
and 60 days. There is a marked loss of statistical power
variability from a small amount of experimental, or random,
more apparent for treatments of lower potency.